BMD plot with color gradient
bmdplotwithgradient.RdProvides an ECDF plot of BMD values with a horizontal color gradient coding, for each item, for the theoretical signal as a function of the dose (concentration). The idea is to display the amplitude and the intensity of the response of each item on the BMD ECDF plot, in addition to the BMD ordered values. This plot is of interest especially when not too much items are presented. To maximize the lisibility of the plot, one can manually pre-select items based on its own criteria (e.g. functional group of interest).
Usage
bmdplotwithgradient(extendedres, BMDtype = c("zSD", "xfold"),
xmin, xmax, y0shift = TRUE, scaling = TRUE,
facetby, facetby2,
shapeby, npoints = 50,
line.size, point.size = 1,
ncol4faceting, limits4colgradient,
lowercol = "darkblue", uppercol = "darkred",
add.label, label.size = 2,
BMD_log_transfo = TRUE)Arguments
- extendedres
the dataframe of results provided by bmdcalc (res) or a subset of this data frame (selected lines). This dataframe can be extended with additional columns coming for example from the functional annotation of items, and some lines can be replicated if their corresponding item has more than one annotation. This extended dataframe must at least contain the column giving the BMD values (
BMD.zSDorBMD.xfolddepending of chosen BMDtype), identification of each curve (id), the columnmodelnaming the fitted model and the values of the parameters (columnsb,c,d,e,f).- BMDtype
The type of BMD to plot,
"zSD"(default choice) or"xfold".- xmin
Optional minimal dose/concentration for definition of the x range.
- xmax
Optional maximal dose/concentration for definition of the x range (can be defined as
max(f$omicdata$dose)withfthe output ofdrcfit()for example).- y0shift
If
TRUE(default choice) for each item the signal is shifted to have the theoretical signal at the control at 0.- scaling
If
TRUE, default choice, for each item the signal is shifted to have the theoretical signal at the control at 0 and scaled by dividing by the maximal absolute signal change (up or down) from the signal at the controlmaxychange.- facetby
optional argument naming the column of
extendedreschosen to split the plot in facets usingggplot2::facet_wrap(no split if omitted).- facetby2
optional argument naming the column of
extendedreschosen as an additional argument to split the plot in facets usingggplot2::facet_grid, with columns defined byfacetbyand rows defined byfacetby2(no split if omitted).- shapeby
optional argument naming the column of
extendedreschosen to shape the BMD points (no difference if shapeby if omitted).- npoints
Number of points computed on each curve in order to define the signal color gradient (= number of doses or concentrations for which the theoretical signal is computed from the fitted model for each item).
- line.size
Size of the horizontal lines for plotting each signal color gradient.
- point.size
Size of the BMD points.
- ncol4faceting
Number of columns for facetting (not used if
facetby2is also provided.- limits4colgradient
Optional vector giving minimal and maximal value of the signal for the color gradient.
- lowercol
Chosen color for the lower values of the signal.
- uppercol
Chosen color for the upper values of the signal.
- add.label
Points are replaced by labels of items if TRUE.
- label.size
Size of labels if add.label is TRUE.
- BMD_log_transfo
If TRUE, the default option, a log transformation of the BMD is used in the plot. This option cannot be used with a null value of xmin in input.
Details
BMD values are plotted as an ECDF plot, as with plot.bmdcalc
using "ecdf" as plottype. In addition is plotted an horizontal color gradient
for each item coding for the signal level at each dose
(or concentration). The optional use of columns to code for shape and/or
facets for each item is particularly intended to give a view of all the dose-response
per group (e.g. metabolic pathways). Those groups must be coded in a column
of extendedres. In case where one item is allocated to more than one group
during the annotation process, the line of this item must be replicated in
extendedres as many times as the number of annotation groups in which it was
allocated.
For each item of the extended dataframe, the name of the model
(column model) and the values of
the parameters (columns b, c, d, e, f)
are used to compute theoretical dose-response curves, and so the
corresponding signal color gradient, in the range [xmin ; xmax].
See also
See plot.bmdcalc and plot.bmdboot.
Examples
# (1)
# A toy example on a very small subsample of a microarray data set
datafilename <- system.file("extdata", "transcripto_very_small_sample.txt", package="DRomics")
o <- microarraydata(datafilename, check = TRUE, norm.method = "cyclicloess")
#> Just wait, the normalization using cyclicloess may take a few minutes.
s_quad <- itemselect(o, select.method = "quadratic", FDR = 0.01)
#> Removing intercept from test coefficients
f <- drcfit(s_quad, progressbar = TRUE)
#> The fitting may be long if the number of selected items is high.
#>
|
| | 0%
|
|==== | 6%
|
|======== | 12%
|
|============ | 18%
|
|================ | 24%
|
|===================== | 29%
|
|========================= | 35%
|
|============================= | 41%
|
|================================= | 47%
|
|===================================== | 53%
|
|========================================= | 59%
|
|============================================= | 65%
|
|================================================= | 71%
|
|====================================================== | 76%
|
|========================================================== | 82%
|
|============================================================== | 88%
|
|================================================================== | 94%
|
|======================================================================| 100%
r <- bmdcalc(f)
# Plot of all the BMD values with color dose-response gradient
#
bmdplotwithgradient(r$res, BMDtype = "zSD")
#> Warning:
#> By default xmax was fixed at the maximal BMD value, but you could also
#> fix it at the maximal tested dose.
#> Warning: Using `size` aesthetic for lines was deprecated in ggplot2 3.4.0.
#> ℹ Please use `linewidth` instead.
#> ℹ The deprecated feature was likely used in the DRomics package.
#> Please report the issue at <https://github.com/lbbe-software/DRomics/issues>.
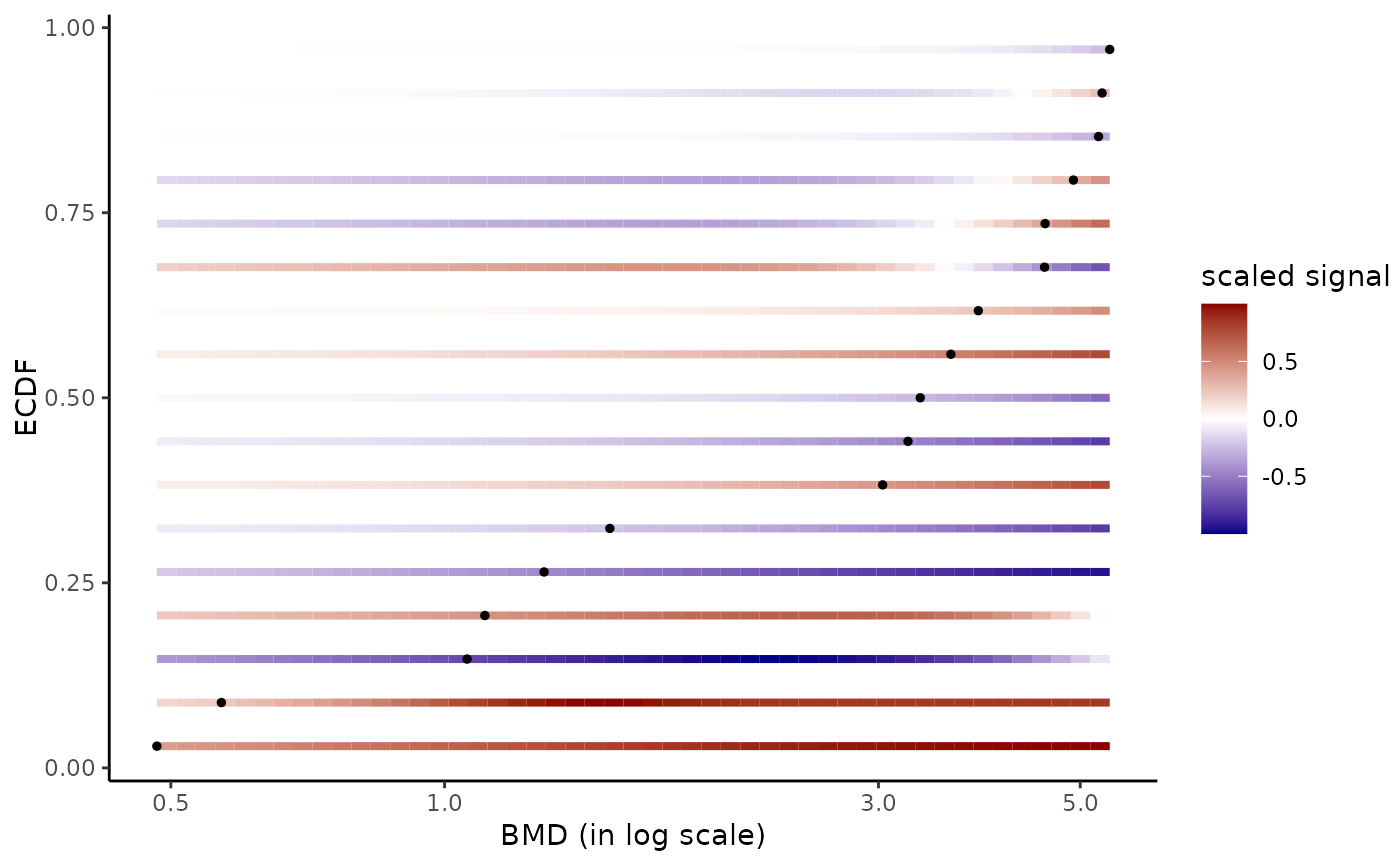 # \donttest{
# Same plot without signal scaling
#
bmdplotwithgradient(r$res, BMDtype = "zSD", scaling = FALSE)
#> Warning:
#> By default xmax was fixed at the maximal BMD value, but you could also
#> fix it at the maximal tested dose.
# \donttest{
# Same plot without signal scaling
#
bmdplotwithgradient(r$res, BMDtype = "zSD", scaling = FALSE)
#> Warning:
#> By default xmax was fixed at the maximal BMD value, but you could also
#> fix it at the maximal tested dose.
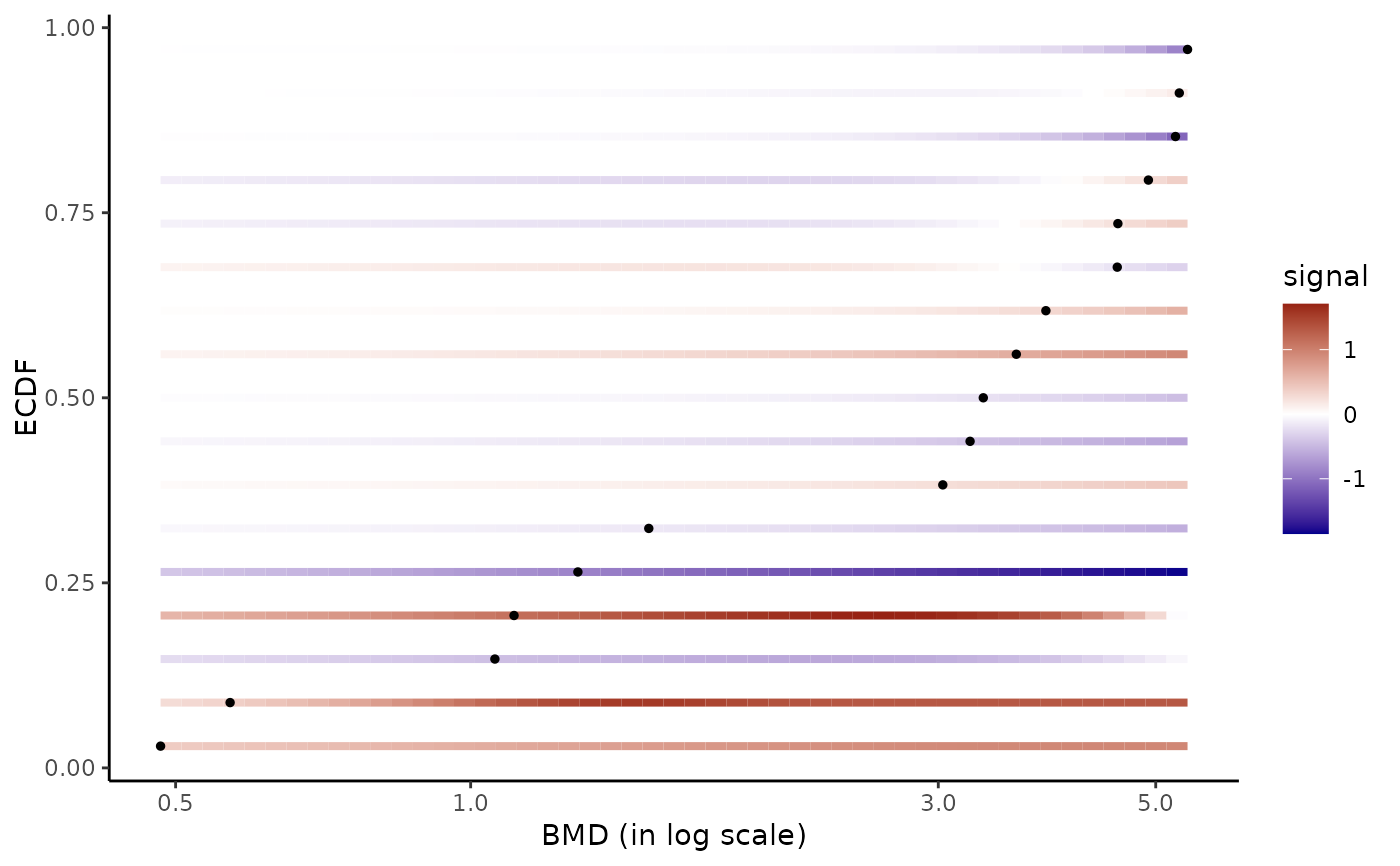 # Plot of all the BMD values with color dose-response gradient
# with definition of xmax from the maximal tested dose
#
bmdplotwithgradient(r$res, BMDtype = "zSD",
xmax = max(f$omicdata$dose))
# Plot of all the BMD values with color dose-response gradient
# with definition of xmax from the maximal tested dose
#
bmdplotwithgradient(r$res, BMDtype = "zSD",
xmax = max(f$omicdata$dose))
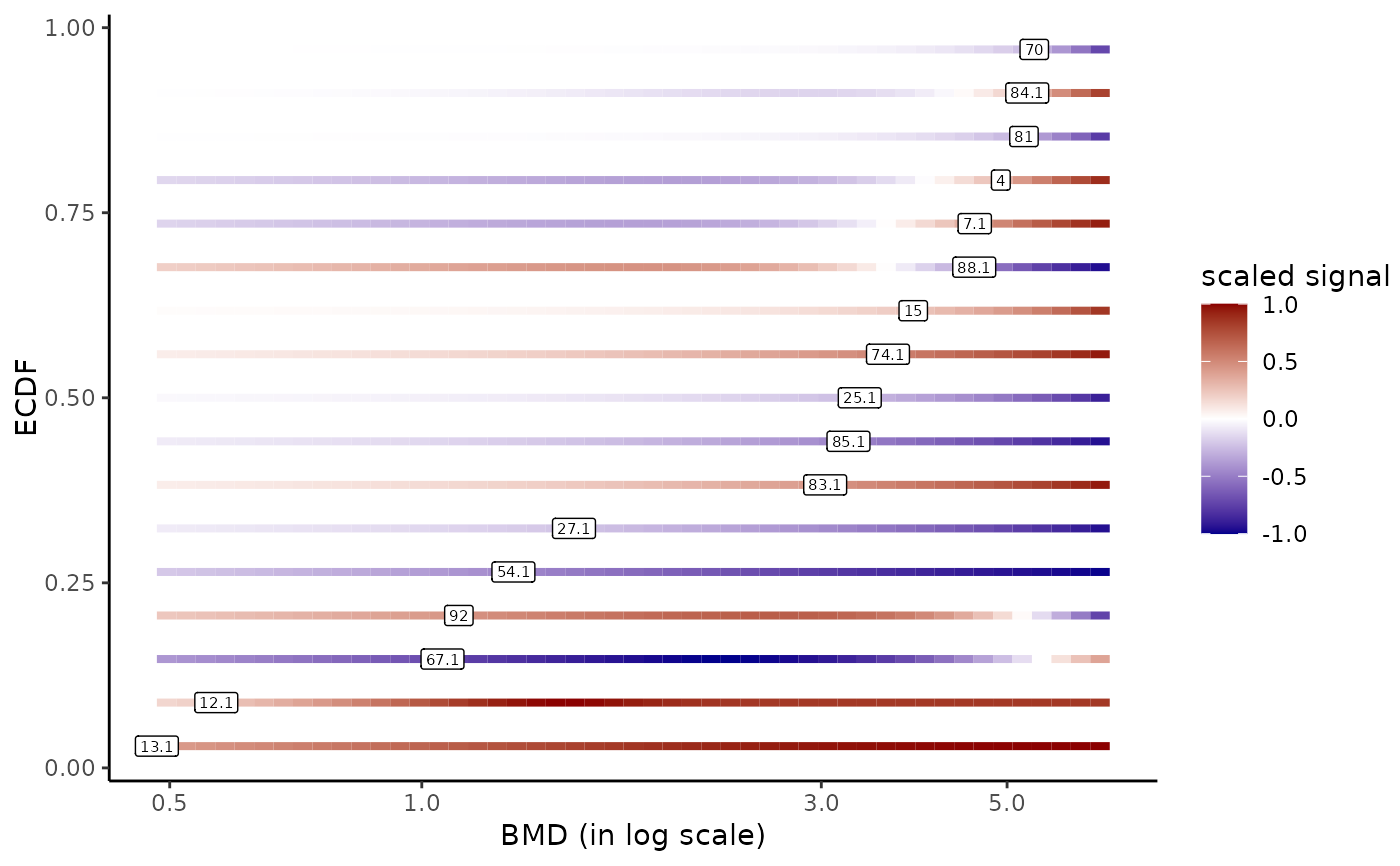
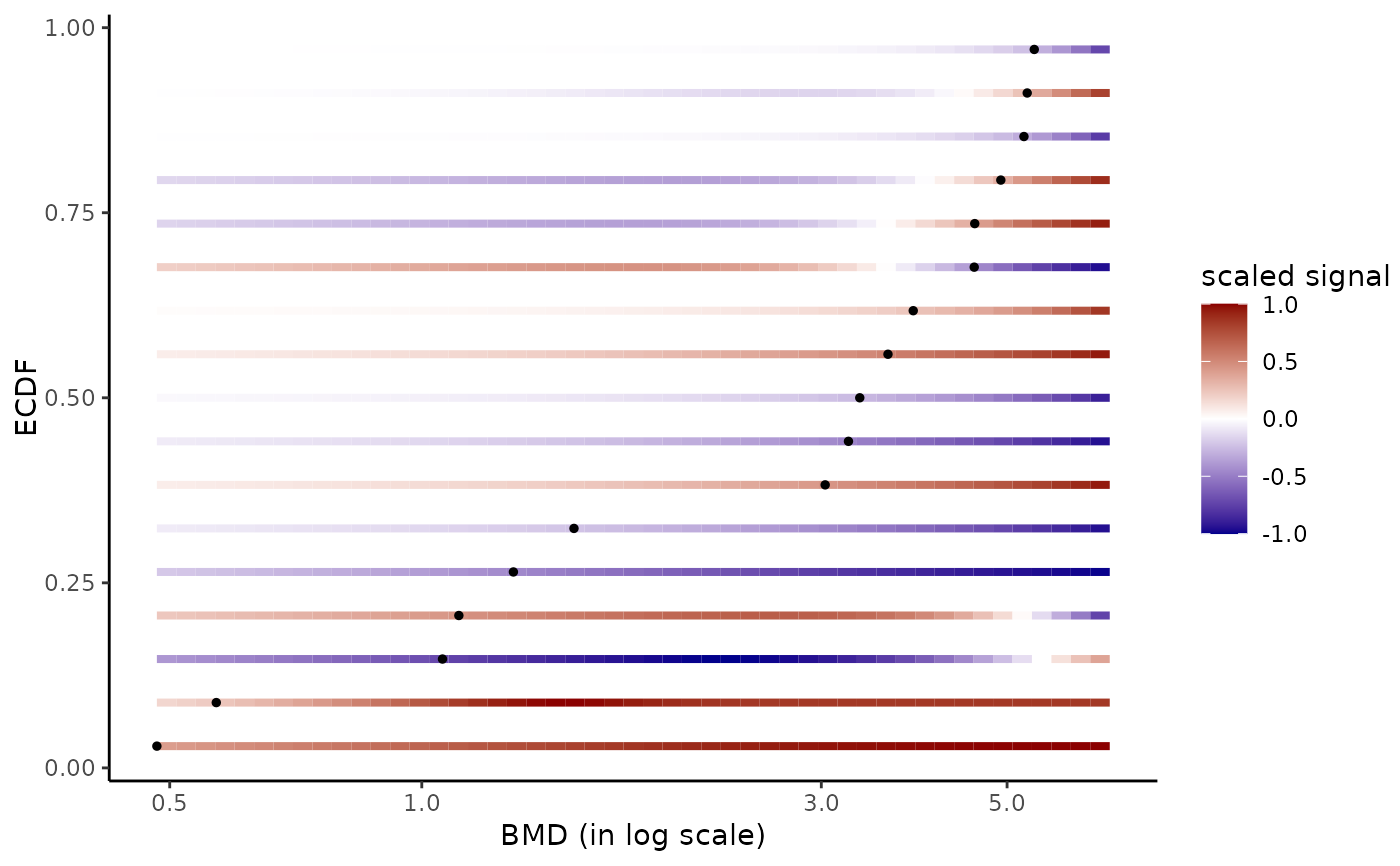 # Add of item labels
bmdplotwithgradient(r$res, BMDtype = "zSD",
xmax = max(f$omicdata$dose), add.label = TRUE)
# Add of item labels
bmdplotwithgradient(r$res, BMDtype = "zSD",
xmax = max(f$omicdata$dose), add.label = TRUE)
 # The same plot in raw scale (we can fix xmin at 0 in this case)
#
bmdplotwithgradient(r$res, BMDtype = "zSD", xmin = 0,
BMD_log_transfo = FALSE)
#> Warning:
#> By default xmax was fixed at the maximal BMD value, but you could also
#> fix it at the maximal tested dose.
# The same plot in raw scale (we can fix xmin at 0 in this case)
#
bmdplotwithgradient(r$res, BMDtype = "zSD", xmin = 0,
BMD_log_transfo = FALSE)
#> Warning:
#> By default xmax was fixed at the maximal BMD value, but you could also
#> fix it at the maximal tested dose.
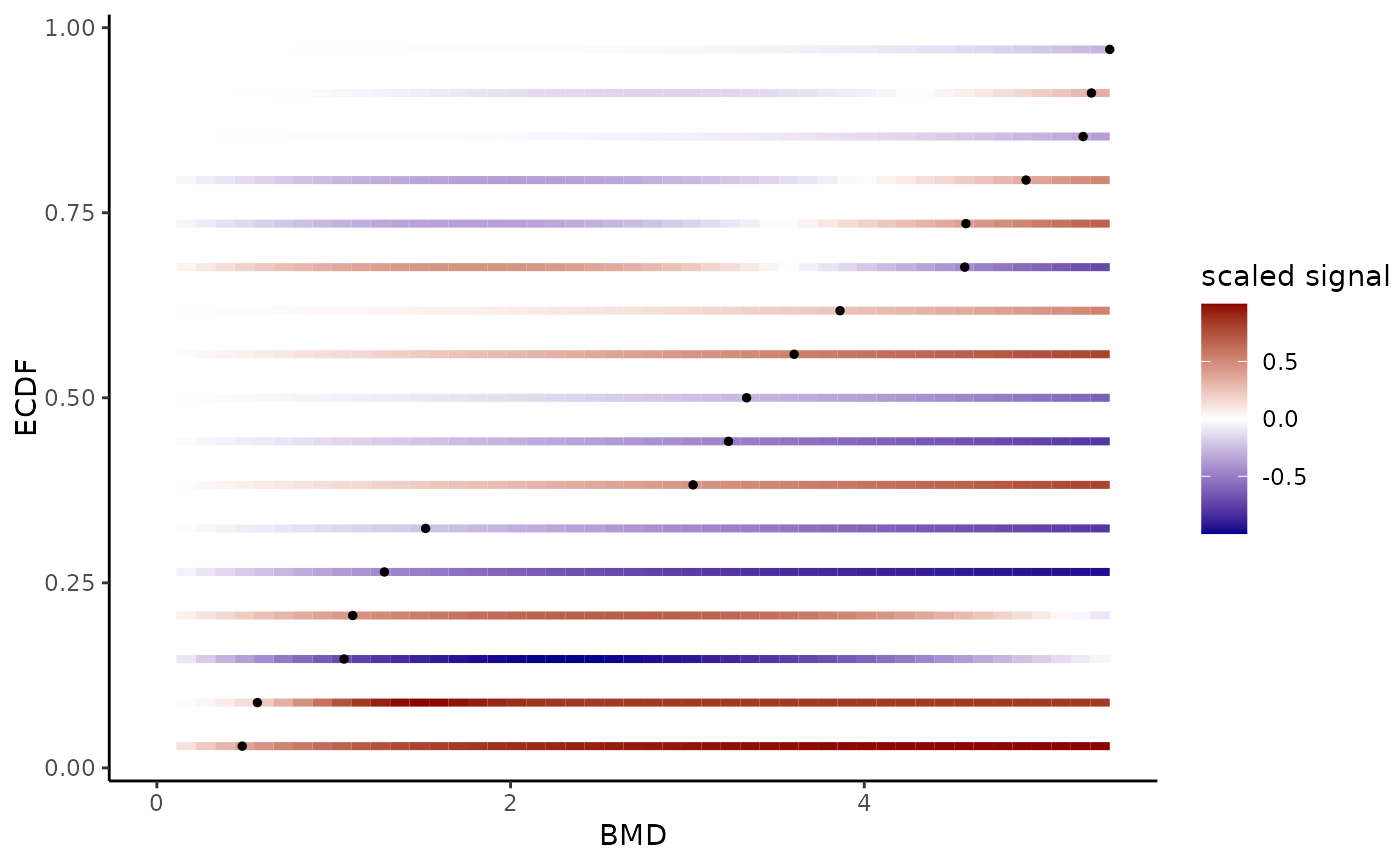 # The same plot in log scale with defining xmin and xmax at a chosen values
#
bmdplotwithgradient(r$res, BMDtype = "zSD",
xmin = min(f$omicdata$dose[f$omicdata$dose != 0] / 2),
xmax = max(f$omicdata$dose),
BMD_log_transfo = TRUE)
# The same plot in log scale with defining xmin and xmax at a chosen values
#
bmdplotwithgradient(r$res, BMDtype = "zSD",
xmin = min(f$omicdata$dose[f$omicdata$dose != 0] / 2),
xmax = max(f$omicdata$dose),
BMD_log_transfo = TRUE)
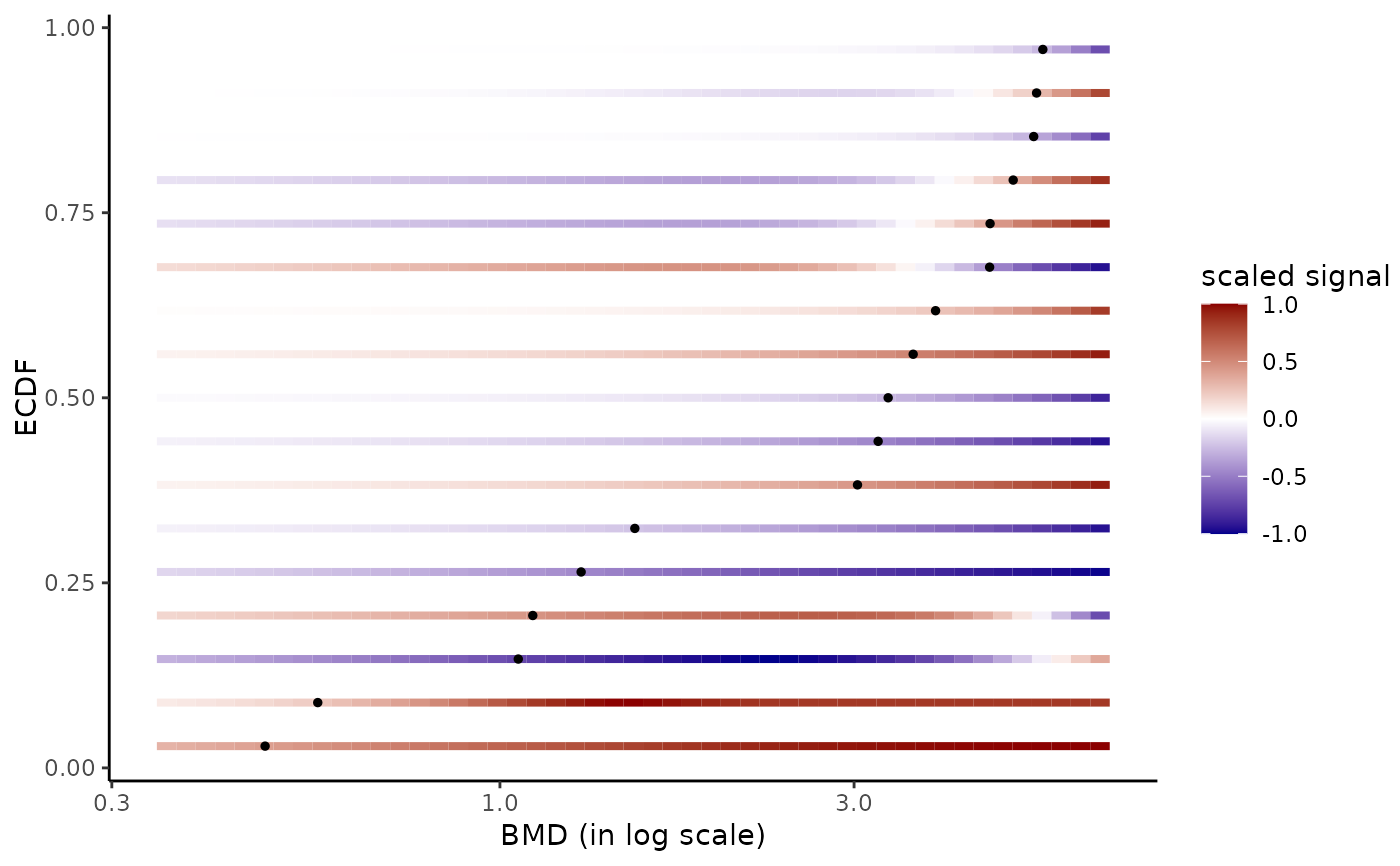 # Plot of all the BMD values with color dose-response gradient
# faceted by response trend and shaped by model
#
bmdplotwithgradient(r$res, BMDtype = "zSD",
facetby = "trend", shapeby = "model")
#> Warning:
#> By default xmax was fixed at the maximal BMD value, but you could also
#> fix it at the maximal tested dose.
# Plot of all the BMD values with color dose-response gradient
# faceted by response trend and shaped by model
#
bmdplotwithgradient(r$res, BMDtype = "zSD",
facetby = "trend", shapeby = "model")
#> Warning:
#> By default xmax was fixed at the maximal BMD value, but you could also
#> fix it at the maximal tested dose.
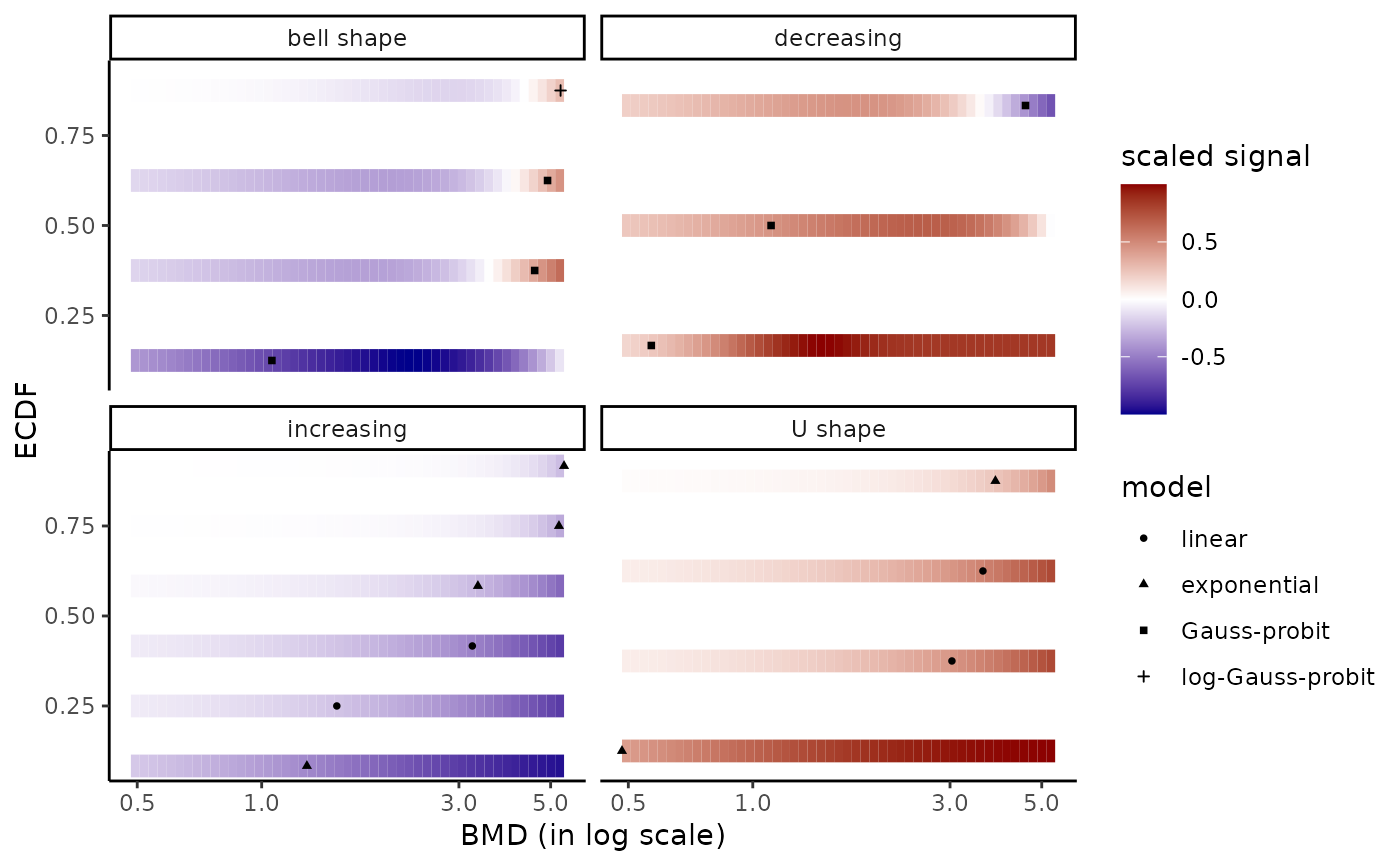
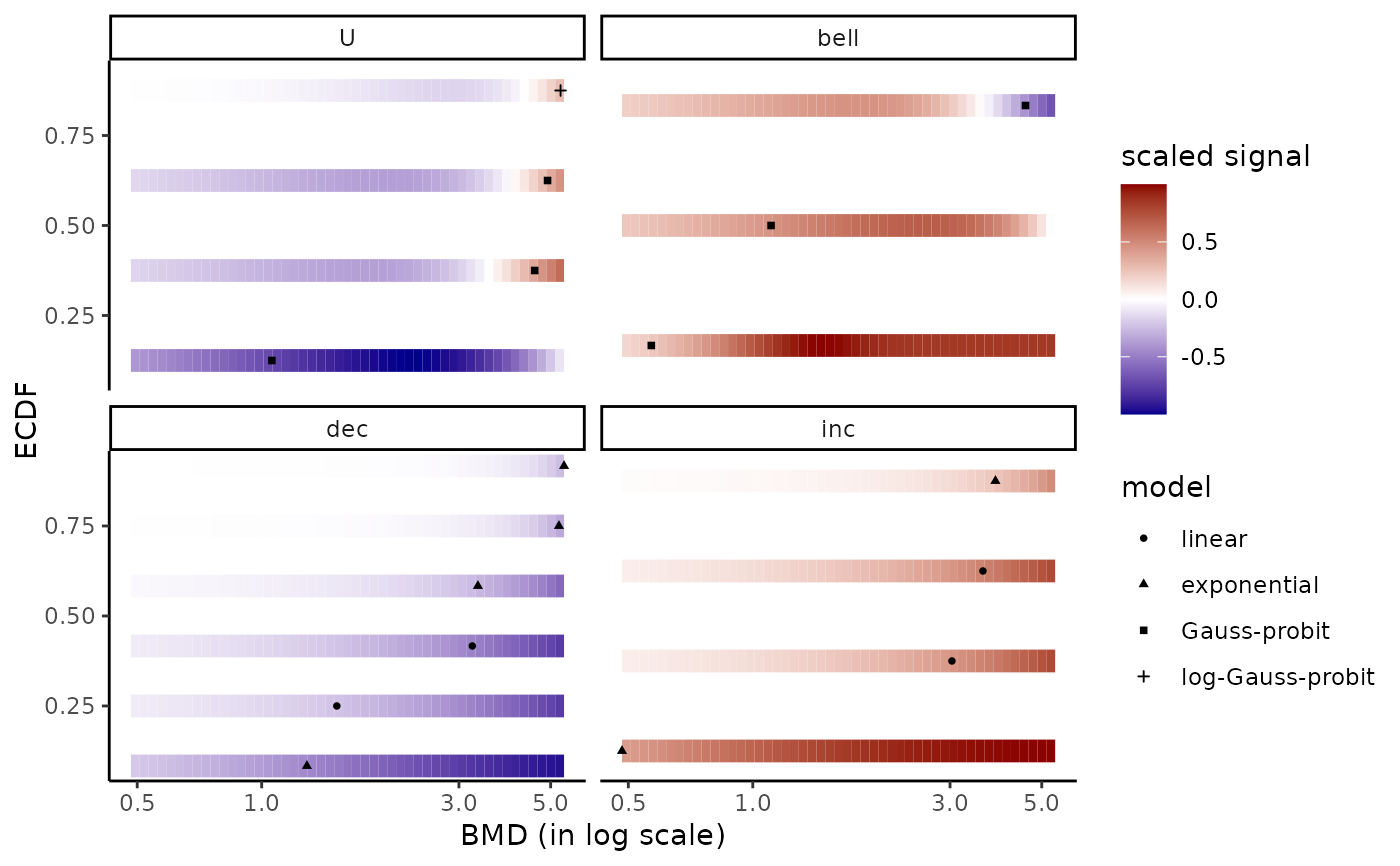 # same plot changing the names of the facets
levels(r$res$trend)
#> [1] "U" "bell" "dec" "inc"
levels(r$res$trend) <- c("bell shape", "decreasing", "increasing", "U shape")
bmdplotwithgradient(r$res, BMDtype = "zSD",
facetby = "trend", shapeby = "model")
#> Warning:
#> By default xmax was fixed at the maximal BMD value, but you could also
#> fix it at the maximal tested dose.
# same plot changing the names of the facets
levels(r$res$trend)
#> [1] "U" "bell" "dec" "inc"
levels(r$res$trend) <- c("bell shape", "decreasing", "increasing", "U shape")
bmdplotwithgradient(r$res, BMDtype = "zSD",
facetby = "trend", shapeby = "model")
#> Warning:
#> By default xmax was fixed at the maximal BMD value, but you could also
#> fix it at the maximal tested dose.
 # same plot changing the labels of the legends
# and inversing the two guides
if (require(ggplot2)) bmdplotwithgradient(r$res, BMDtype = "zSD",
facetby = "trend", shapeby = "model") +
labs(col = "signal value", shape = "model") +
guides(colour = guide_colourbar(order = 1),
shape = guide_legend(order = 2))
#> Loading required package: ggplot2
#> Warning:
#> By default xmax was fixed at the maximal BMD value, but you could also
#> fix it at the maximal tested dose.
# same plot changing the labels of the legends
# and inversing the two guides
if (require(ggplot2)) bmdplotwithgradient(r$res, BMDtype = "zSD",
facetby = "trend", shapeby = "model") +
labs(col = "signal value", shape = "model") +
guides(colour = guide_colourbar(order = 1),
shape = guide_legend(order = 2))
#> Loading required package: ggplot2
#> Warning:
#> By default xmax was fixed at the maximal BMD value, but you could also
#> fix it at the maximal tested dose.
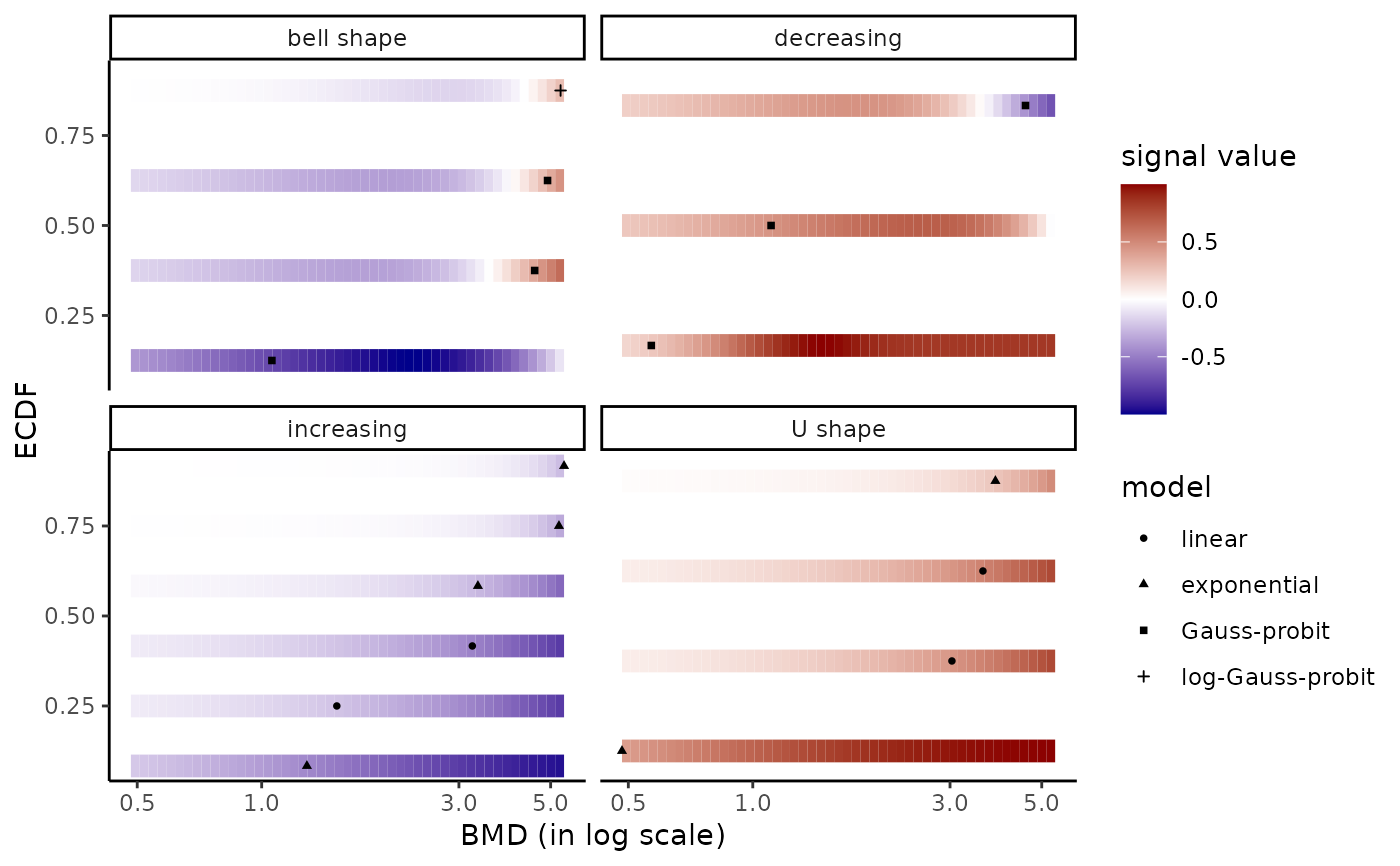 # (2)
# Plot of BMD values with color dose-response gradient
# faceted by metabolic pathway (from annotation of the selected items)
# and shaped by dose-response trend
# An example from the paper published by Larras et al. 2020
# in Journal of Hazardous Materials
# https://doi.org/10.1016/j.jhazmat.2020.122727
# A example of plot obtained with this function is in Figure 5 in Larras et al. 2020
# the dataframe with metabolomic results (output $res of bmdcalc() or bmdboot() functions)
resfilename <- system.file("extdata", "triclosanSVmetabres.txt", package="DRomics")
res <- read.table(resfilename, header = TRUE, stringsAsFactors = TRUE)
str(res)
#> 'data.frame': 31 obs. of 27 variables:
#> $ id : Factor w/ 31 levels "NAP47_51","NAP_2",..: 2 3 4 5 6 7 8 9 10 11 ...
#> $ irow : int 2 21 28 34 38 47 49 51 53 67 ...
#> $ adjpvalue : num 6.23e-05 1.11e-05 1.03e-05 1.89e-03 4.16e-03 ...
#> $ model : Factor w/ 4 levels "Gauss-probit",..: 2 3 3 2 2 4 2 2 3 3 ...
#> $ nbpar : int 3 2 2 3 3 5 3 3 2 2 ...
#> $ b : num 0.4598 -0.0595 -0.0451 0.6011 0.6721 ...
#> $ c : num NA NA NA NA NA ...
#> $ d : num 5.94 5.39 7.86 6.86 6.21 ...
#> $ e : num -1.648 NA NA -0.321 -0.323 ...
#> $ f : num NA NA NA NA NA ...
#> $ SDres : num 0.126 0.0793 0.052 0.2338 0.2897 ...
#> $ typology : Factor w/ 10 levels "E.dec.concave",..: 2 7 7 2 2 9 2 2 7 7 ...
#> $ trend : Factor w/ 4 levels "U","bell","dec",..: 3 3 3 3 3 1 3 3 3 3 ...
#> $ y0 : num 5.94 5.39 7.86 6.86 6.21 ...
#> $ yrange : num 0.456 0.461 0.35 0.601 0.672 ...
#> $ maxychange : num 0.456 0.461 0.35 0.601 0.672 ...
#> $ xextrem : num NA NA NA NA NA ...
#> $ yextrem : num NA NA NA NA NA ...
#> $ BMD.zSD : num 0.528 1.333 1.154 0.158 0.182 ...
#> $ BMR.zSD : num 5.82 5.31 7.81 6.62 5.92 ...
#> $ BMD.xfold : num NA NA NA NA 0.832 ...
#> $ BMR.xfold : num 5.35 4.85 7.07 6.17 5.59 ...
#> $ BMD.zSD.lower : num 0.2001 0.8534 0.7519 0.0554 0.081 ...
#> $ BMD.zSD.upper : num 1.11 1.746 1.465 0.68 0.794 ...
#> $ BMD.xfold.lower : num Inf 7.611 Inf 0.561 0.329 ...
#> $ BMD.xfold.upper : num Inf Inf Inf Inf Inf ...
#> $ nboot.successful: int 957 1000 1000 648 620 872 909 565 1000 1000 ...
# the dataframe with annotation of each item identified in the previous file
# each item may have more than one annotation (-> more than one line)
annotfilename <- system.file("extdata", "triclosanSVmetabannot.txt", package="DRomics")
annot <- read.table(annotfilename, header = TRUE, stringsAsFactors = TRUE)
str(annot)
#> 'data.frame': 84 obs. of 2 variables:
#> $ metab.code: Factor w/ 31 levels "NAP47_51","NAP_2",..: 2 3 4 4 4 4 5 6 7 8 ...
#> $ path_class: Factor w/ 9 levels "Amino acid metabolism",..: 5 3 3 2 6 8 5 5 5 5 ...
# Merging of both previous dataframes
# in order to obtain an extenderes dataframe
extendedres <- merge(x = res, y = annot, by.x = "id", by.y = "metab.code")
head(extendedres)
#> id irow adjpvalue model nbpar b c d
#> 1 NAP47_51 46 7.158246e-04 linear 2 -0.05600559 NA 7.343571
#> 2 NAP_2 2 6.232579e-05 exponential 3 0.45981242 NA 5.941896
#> 3 NAP_23 21 1.106958e-05 linear 2 -0.05946618 NA 5.387252
#> 4 NAP_30 28 1.028343e-05 linear 2 -0.04507832 NA 7.859109
#> 5 NAP_30 28 1.028343e-05 linear 2 -0.04507832 NA 7.859109
#> 6 NAP_30 28 1.028343e-05 linear 2 -0.04507832 NA 7.859109
#> e f SDres typology trend y0 yrange maxychange
#> 1 NA NA 0.12454183 L.dec dec 7.343571 0.4346034 0.4346034
#> 2 -1.647958 NA 0.12604568 E.dec.convex dec 5.941896 0.4556672 0.4556672
#> 3 NA NA 0.07929266 L.dec dec 5.387252 0.4614576 0.4614576
#> 4 NA NA 0.05203245 L.dec dec 7.859109 0.3498078 0.3498078
#> 5 NA NA 0.05203245 L.dec dec 7.859109 0.3498078 0.3498078
#> 6 NA NA 0.05203245 L.dec dec 7.859109 0.3498078 0.3498078
#> xextrem yextrem BMD.zSD BMR.zSD BMD.xfold BMR.xfold BMD.zSD.lower
#> 1 NA NA 2.2237393 7.219029 NA 6.609214 0.9785095
#> 2 NA NA 0.5279668 5.815850 NA 5.347706 0.2000881
#> 3 NA NA 1.3334076 5.307960 NA 4.848527 0.8533711
#> 4 NA NA 1.1542677 7.807077 NA 7.073198 0.7518588
#> 5 NA NA 1.1542677 7.807077 NA 7.073198 0.7518588
#> 6 NA NA 1.1542677 7.807077 NA 7.073198 0.7518588
#> BMD.zSD.upper BMD.xfold.lower BMD.xfold.upper nboot.successful
#> 1 4.068699 Inf Inf 1000
#> 2 1.109559 Inf Inf 957
#> 3 1.746010 7.610936 Inf 1000
#> 4 1.464998 Inf Inf 1000
#> 5 1.464998 Inf Inf 1000
#> 6 1.464998 Inf Inf 1000
#> path_class
#> 1 Lipid metabolism
#> 2 Lipid metabolism
#> 3 Carbohydrate metabolism
#> 4 Carbohydrate metabolism
#> 5 Biosynthesis of other secondary metabolites
#> 6 Membrane transport
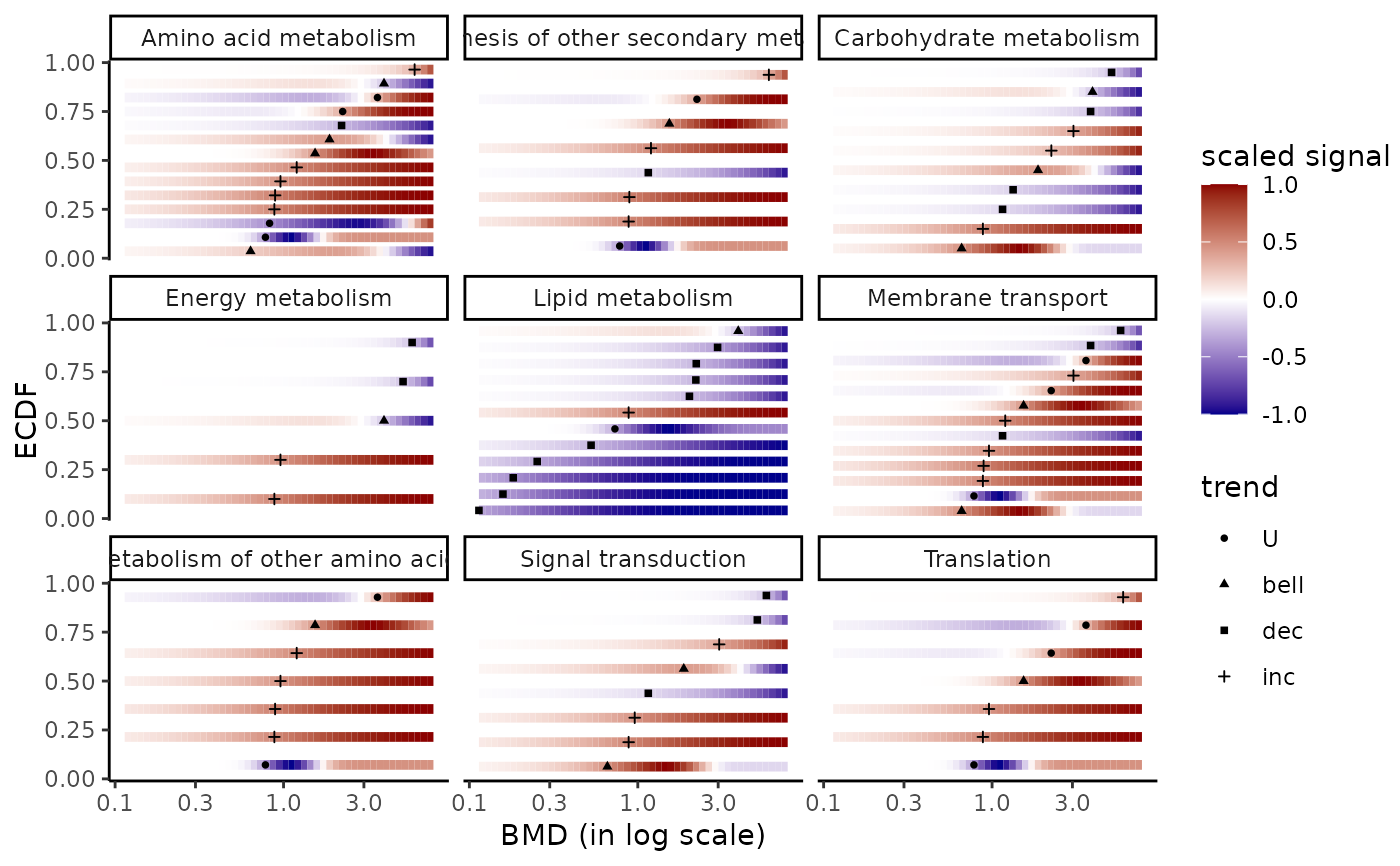
### (2.a) BMDplot with gradient by pathway
bmdplotwithgradient(extendedres, BMDtype = "zSD",
facetby = "path_class",
xmax = 7.76, # maximal tested dose in those data
shapeby = "trend")
# (2)
# Plot of BMD values with color dose-response gradient
# faceted by metabolic pathway (from annotation of the selected items)
# and shaped by dose-response trend
# An example from the paper published by Larras et al. 2020
# in Journal of Hazardous Materials
# https://doi.org/10.1016/j.jhazmat.2020.122727
# A example of plot obtained with this function is in Figure 5 in Larras et al. 2020
# the dataframe with metabolomic results (output $res of bmdcalc() or bmdboot() functions)
resfilename <- system.file("extdata", "triclosanSVmetabres.txt", package="DRomics")
res <- read.table(resfilename, header = TRUE, stringsAsFactors = TRUE)
str(res)
#> 'data.frame': 31 obs. of 27 variables:
#> $ id : Factor w/ 31 levels "NAP47_51","NAP_2",..: 2 3 4 5 6 7 8 9 10 11 ...
#> $ irow : int 2 21 28 34 38 47 49 51 53 67 ...
#> $ adjpvalue : num 6.23e-05 1.11e-05 1.03e-05 1.89e-03 4.16e-03 ...
#> $ model : Factor w/ 4 levels "Gauss-probit",..: 2 3 3 2 2 4 2 2 3 3 ...
#> $ nbpar : int 3 2 2 3 3 5 3 3 2 2 ...
#> $ b : num 0.4598 -0.0595 -0.0451 0.6011 0.6721 ...
#> $ c : num NA NA NA NA NA ...
#> $ d : num 5.94 5.39 7.86 6.86 6.21 ...
#> $ e : num -1.648 NA NA -0.321 -0.323 ...
#> $ f : num NA NA NA NA NA ...
#> $ SDres : num 0.126 0.0793 0.052 0.2338 0.2897 ...
#> $ typology : Factor w/ 10 levels "E.dec.concave",..: 2 7 7 2 2 9 2 2 7 7 ...
#> $ trend : Factor w/ 4 levels "U","bell","dec",..: 3 3 3 3 3 1 3 3 3 3 ...
#> $ y0 : num 5.94 5.39 7.86 6.86 6.21 ...
#> $ yrange : num 0.456 0.461 0.35 0.601 0.672 ...
#> $ maxychange : num 0.456 0.461 0.35 0.601 0.672 ...
#> $ xextrem : num NA NA NA NA NA ...
#> $ yextrem : num NA NA NA NA NA ...
#> $ BMD.zSD : num 0.528 1.333 1.154 0.158 0.182 ...
#> $ BMR.zSD : num 5.82 5.31 7.81 6.62 5.92 ...
#> $ BMD.xfold : num NA NA NA NA 0.832 ...
#> $ BMR.xfold : num 5.35 4.85 7.07 6.17 5.59 ...
#> $ BMD.zSD.lower : num 0.2001 0.8534 0.7519 0.0554 0.081 ...
#> $ BMD.zSD.upper : num 1.11 1.746 1.465 0.68 0.794 ...
#> $ BMD.xfold.lower : num Inf 7.611 Inf 0.561 0.329 ...
#> $ BMD.xfold.upper : num Inf Inf Inf Inf Inf ...
#> $ nboot.successful: int 957 1000 1000 648 620 872 909 565 1000 1000 ...
# the dataframe with annotation of each item identified in the previous file
# each item may have more than one annotation (-> more than one line)
annotfilename <- system.file("extdata", "triclosanSVmetabannot.txt", package="DRomics")
annot <- read.table(annotfilename, header = TRUE, stringsAsFactors = TRUE)
str(annot)
#> 'data.frame': 84 obs. of 2 variables:
#> $ metab.code: Factor w/ 31 levels "NAP47_51","NAP_2",..: 2 3 4 4 4 4 5 6 7 8 ...
#> $ path_class: Factor w/ 9 levels "Amino acid metabolism",..: 5 3 3 2 6 8 5 5 5 5 ...
# Merging of both previous dataframes
# in order to obtain an extenderes dataframe
extendedres <- merge(x = res, y = annot, by.x = "id", by.y = "metab.code")
head(extendedres)
#> id irow adjpvalue model nbpar b c d
#> 1 NAP47_51 46 7.158246e-04 linear 2 -0.05600559 NA 7.343571
#> 2 NAP_2 2 6.232579e-05 exponential 3 0.45981242 NA 5.941896
#> 3 NAP_23 21 1.106958e-05 linear 2 -0.05946618 NA 5.387252
#> 4 NAP_30 28 1.028343e-05 linear 2 -0.04507832 NA 7.859109
#> 5 NAP_30 28 1.028343e-05 linear 2 -0.04507832 NA 7.859109
#> 6 NAP_30 28 1.028343e-05 linear 2 -0.04507832 NA 7.859109
#> e f SDres typology trend y0 yrange maxychange
#> 1 NA NA 0.12454183 L.dec dec 7.343571 0.4346034 0.4346034
#> 2 -1.647958 NA 0.12604568 E.dec.convex dec 5.941896 0.4556672 0.4556672
#> 3 NA NA 0.07929266 L.dec dec 5.387252 0.4614576 0.4614576
#> 4 NA NA 0.05203245 L.dec dec 7.859109 0.3498078 0.3498078
#> 5 NA NA 0.05203245 L.dec dec 7.859109 0.3498078 0.3498078
#> 6 NA NA 0.05203245 L.dec dec 7.859109 0.3498078 0.3498078
#> xextrem yextrem BMD.zSD BMR.zSD BMD.xfold BMR.xfold BMD.zSD.lower
#> 1 NA NA 2.2237393 7.219029 NA 6.609214 0.9785095
#> 2 NA NA 0.5279668 5.815850 NA 5.347706 0.2000881
#> 3 NA NA 1.3334076 5.307960 NA 4.848527 0.8533711
#> 4 NA NA 1.1542677 7.807077 NA 7.073198 0.7518588
#> 5 NA NA 1.1542677 7.807077 NA 7.073198 0.7518588
#> 6 NA NA 1.1542677 7.807077 NA 7.073198 0.7518588
#> BMD.zSD.upper BMD.xfold.lower BMD.xfold.upper nboot.successful
#> 1 4.068699 Inf Inf 1000
#> 2 1.109559 Inf Inf 957
#> 3 1.746010 7.610936 Inf 1000
#> 4 1.464998 Inf Inf 1000
#> 5 1.464998 Inf Inf 1000
#> 6 1.464998 Inf Inf 1000
#> path_class
#> 1 Lipid metabolism
#> 2 Lipid metabolism
#> 3 Carbohydrate metabolism
#> 4 Carbohydrate metabolism
#> 5 Biosynthesis of other secondary metabolites
#> 6 Membrane transport
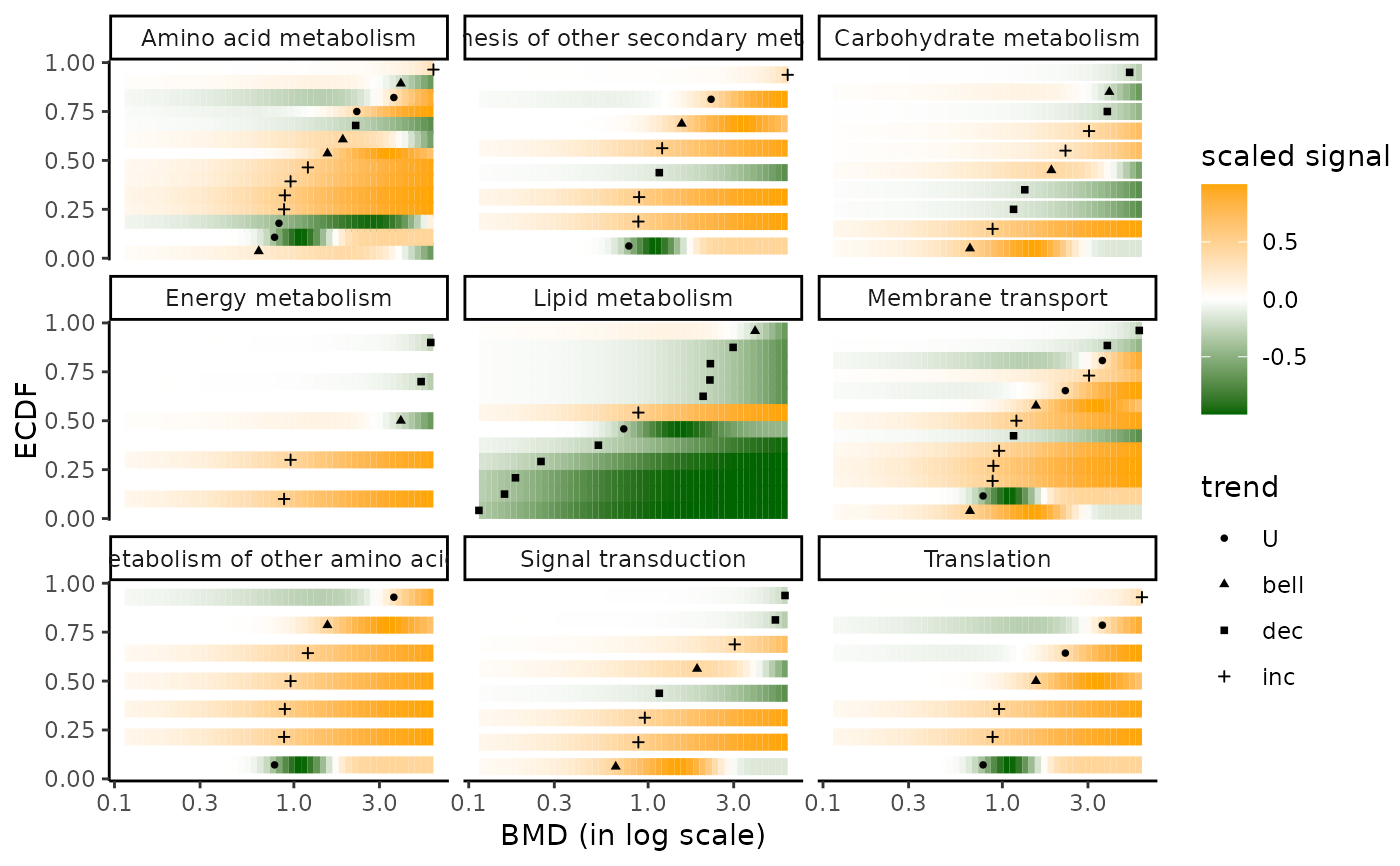
### (2.a) BMDplot with gradient by pathway
bmdplotwithgradient(extendedres, BMDtype = "zSD",
facetby = "path_class",
xmax = 7.76, # maximal tested dose in those data
shapeby = "trend")
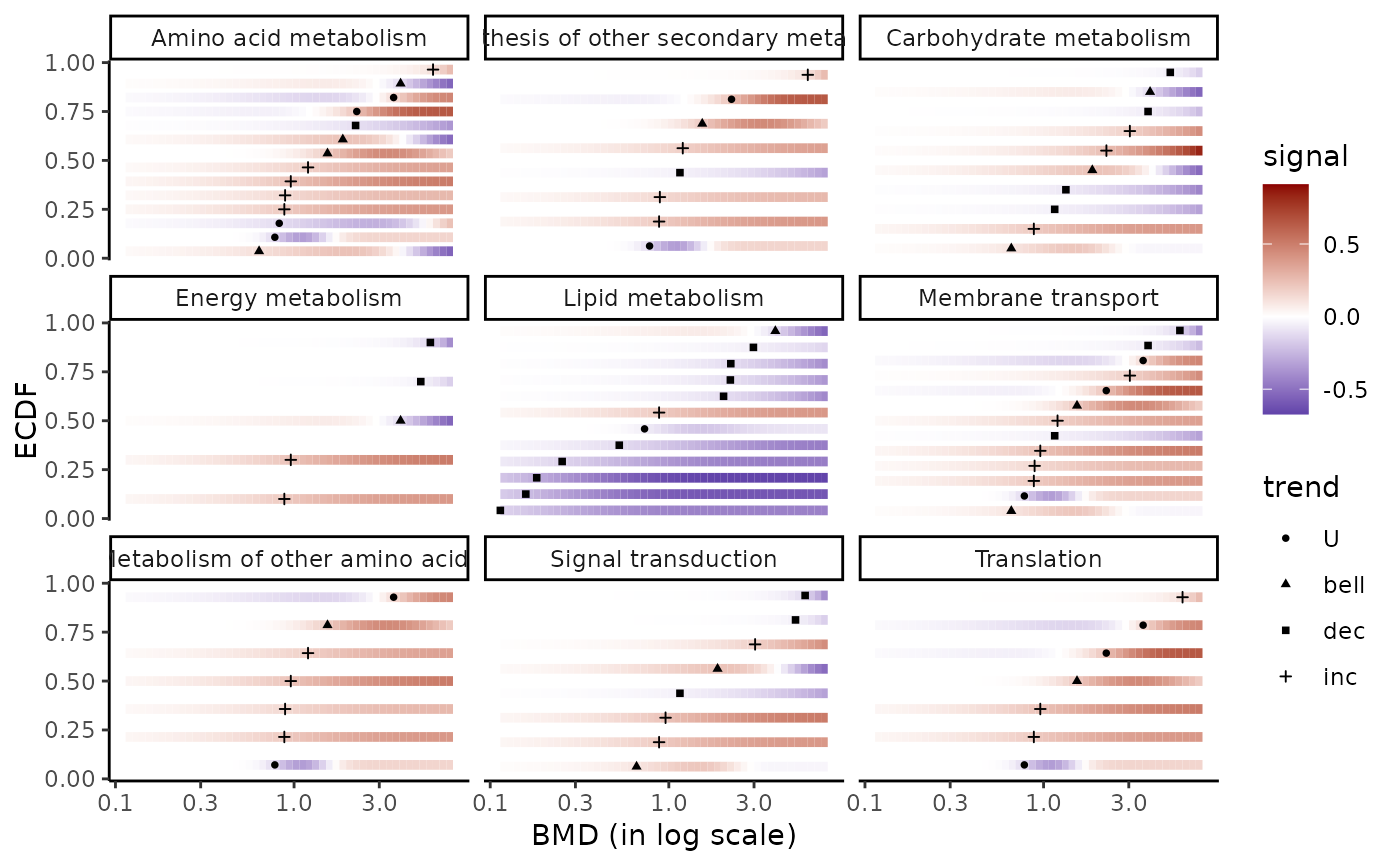
 ### (2.a) BMDplot with gradient by pathway without scaling
bmdplotwithgradient(extendedres, BMDtype = "zSD",
facetby = "path_class", xmax = 7.76,
shapeby = "trend", scaling = FALSE)
### (2.a) BMDplot with gradient by pathway without scaling
bmdplotwithgradient(extendedres, BMDtype = "zSD",
facetby = "path_class", xmax = 7.76,
shapeby = "trend", scaling = FALSE)
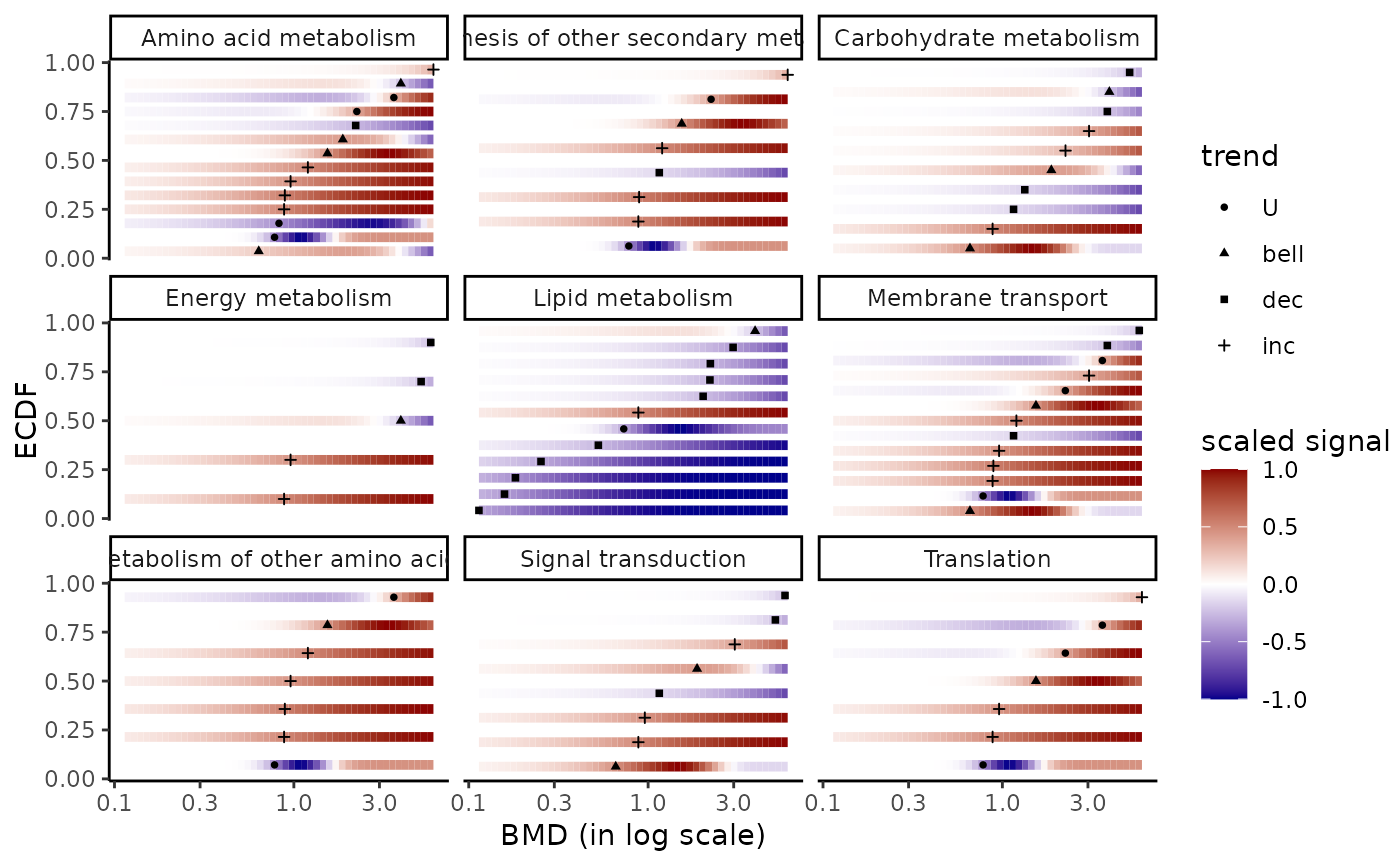
 # (2.b) BMDplot with gradient by pathway
# forcing the limits of the colour gradient at other
# values than observed minimal and maximal values of the signal
bmdplotwithgradient(extendedres, BMDtype = "zSD",
facetby = "path_class",
shapeby = "trend",
limits4colgradient = c(-1, 1))
#> Warning:
#> By default xmax was fixed at the maximal BMD value, but you could also
#> fix it at the maximal tested dose.
# (2.b) BMDplot with gradient by pathway
# forcing the limits of the colour gradient at other
# values than observed minimal and maximal values of the signal
bmdplotwithgradient(extendedres, BMDtype = "zSD",
facetby = "path_class",
shapeby = "trend",
limits4colgradient = c(-1, 1))
#> Warning:
#> By default xmax was fixed at the maximal BMD value, but you could also
#> fix it at the maximal tested dose.
 # (2.c) The same example changing the gradient colors and the line size
bmdplotwithgradient(extendedres, BMDtype = "zSD",
facetby = "path_class",
shapeby = "trend",
line.size = 3,
lowercol = "darkgreen", uppercol = "orange")
#> Warning:
#> By default xmax was fixed at the maximal BMD value, but you could also
#> fix it at the maximal tested dose.
# (2.c) The same example changing the gradient colors and the line size
bmdplotwithgradient(extendedres, BMDtype = "zSD",
facetby = "path_class",
shapeby = "trend",
line.size = 3,
lowercol = "darkgreen", uppercol = "orange")
#> Warning:
#> By default xmax was fixed at the maximal BMD value, but you could also
#> fix it at the maximal tested dose.
 # (2.d) The same example with only lipid metabolism pathclass
# and identification of the metabolites
LMres <- extendedres[extendedres$path_class == "Lipid metabolism", ]
bmdplotwithgradient(LMres, BMDtype = "zSD",
line.size = 3,
add.label = TRUE, label.size = 3)
#> Warning:
#> By default xmax was fixed at the maximal BMD value, but you could also
#> fix it at the maximal tested dose.
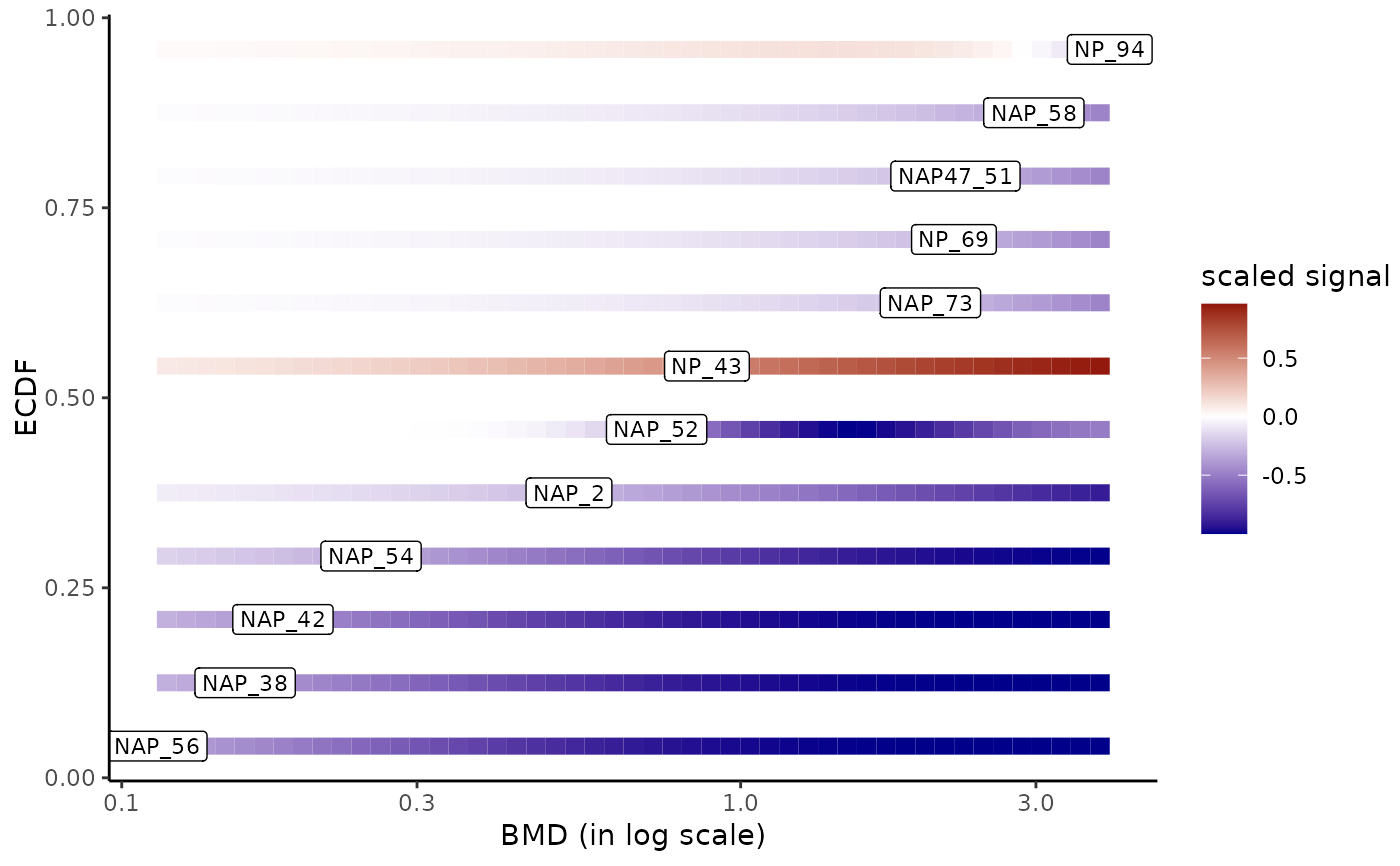
# (2.d) The same example with only lipid metabolism pathclass
# and identification of the metabolites
LMres <- extendedres[extendedres$path_class == "Lipid metabolism", ]
bmdplotwithgradient(LMres, BMDtype = "zSD",
line.size = 3,
add.label = TRUE, label.size = 3)
#> Warning:
#> By default xmax was fixed at the maximal BMD value, but you could also
#> fix it at the maximal tested dose.
 # (3)
# An example on a microarray data set (a subsample of a greater data set)
#
datafilename <- system.file("extdata", "transcripto_sample.txt", package="DRomics")
(o <- microarraydata(datafilename, check = TRUE, norm.method = "cyclicloess"))
#> Just wait, the normalization using cyclicloess may take a few minutes.
#> Elements of the experimental design in order to check the coding of the data:
#> Tested doses and number of replicates for each dose:
#>
#> 0 0.69 1.223 2.148 3.774 6.631
#> 5 5 5 5 5 5
#> Number of items: 1000
#> Identifiers of the first 20 items:
#> [1] "1" "2" "3" "4" "5.1" "5.2" "6.1" "6.2" "7.1" "7.2"
#> [11] "8.1" "8.2" "9.1" "9.2" "10.1" "10.2" "11.1" "11.2" "12.1" "12.2"
#> Data were normalized between arrays using the following method: cyclicloess
(s_quad <- itemselect(o, select.method = "quadratic", FDR = 0.001))
#> Removing intercept from test coefficients
#> Number of selected items using a quadratic trend test with an FDR of 0.001: 78
#> Identifiers of the first 20 most responsive items:
#> [1] "384.2" "383.1" "383.2" "384.1" "301.1" "363.1" "300.2" "364.2" "364.1"
#> [10] "363.2" "301.2" "300.1" "351.1" "350.2" "239.1" "240.1" "240.2" "370"
#> [19] "15" "350.1"
(f <- drcfit(s_quad, progressbar = TRUE))
#> The fitting may be long if the number of selected items is high.
#>
|
| | 0%
|
|= | 1%
|
|== | 3%
|
|=== | 4%
|
|==== | 5%
|
|==== | 6%
|
|===== | 8%
|
|====== | 9%
|
|======= | 10%
|
|======== | 12%
|
|========= | 13%
|
|========== | 14%
|
|=========== | 15%
|
|============ | 17%
|
|============= | 18%
|
|============= | 19%
|
|============== | 21%
|
|=============== | 22%
|
|================ | 23%
|
|================= | 24%
|
|================== | 26%
|
|=================== | 27%
|
|==================== | 28%
|
|===================== | 29%
|
|====================== | 31%
|
|====================== | 32%
|
|======================= | 33%
|
|======================== | 35%
|
|========================= | 36%
|
|========================== | 37%
|
|=========================== | 38%
|
|============================ | 40%
|
|============================= | 41%
|
|============================== | 42%
|
|=============================== | 44%
|
|=============================== | 45%
|
|================================ | 46%
|
|================================= | 47%
|
|================================== | 49%
|
|=================================== | 50%
|
|==================================== | 51%
|
|===================================== | 53%
|
|====================================== | 54%
|
|======================================= | 55%
|
|======================================= | 56%
|
|======================================== | 58%
|
|========================================= | 59%
|
|========================================== | 60%
|
|=========================================== | 62%
|
|============================================ | 63%
|
|============================================= | 64%
|
|============================================== | 65%
|
|=============================================== | 67%
|
|================================================ | 68%
|
|================================================ | 69%
|
|================================================= | 71%
|
|================================================== | 72%
|
|=================================================== | 73%
|
|==================================================== | 74%
|
|===================================================== | 76%
|
|====================================================== | 77%
|
|======================================================= | 78%
|
|======================================================== | 79%
|
|========================================================= | 81%
|
|========================================================= | 82%
|
|========================================================== | 83%
|
|=========================================================== | 85%
|
|============================================================ | 86%
|
|============================================================= | 87%
|
|============================================================== | 88%
|
|=============================================================== | 90%
|
|================================================================ | 91%
|
|================================================================= | 92%
|
|================================================================== | 94%
|
|================================================================== | 95%
|
|=================================================================== | 96%
|
|==================================================================== | 97%
|
|===================================================================== | 99%
|
|======================================================================| 100%
#> Results of the fitting using the AICc to select the best fit model
#> 11 dose-response curves out of 78 previously selected were removed
#> because no model could be fitted reliably.
#> Distribution of the chosen models among the 67 fitted dose-response curves:
#>
#> Hill linear exponential Gauss-probit
#> 0 11 30 23
#> log-Gauss-probit
#> 3
#> Distribution of the trends (curve shapes) among the 67 fitted dose-response curves:
#>
#> U bell dec inc
#> 6 20 22 19
(r <- bmdcalc(f))
#> 1 BMD-xfold values and 0 BMD-zSD values are not defined (coded NaN as
#> the BMR stands outside the range of response values defined by the model).
#> 28 BMD-xfold values and 0 BMD-zSD values could not be calculated (coded
#> NA as the BMR stands within the range of response values defined by the
#> model but outside the range of tested doses).
bmdplotwithgradient(r$res, BMDtype = "zSD",
facetby = "trend",
shapeby = "model")
#> Warning:
#> By default xmax was fixed at the maximal BMD value, but you could also
#> fix it at the maximal tested dose.
# (3)
# An example on a microarray data set (a subsample of a greater data set)
#
datafilename <- system.file("extdata", "transcripto_sample.txt", package="DRomics")
(o <- microarraydata(datafilename, check = TRUE, norm.method = "cyclicloess"))
#> Just wait, the normalization using cyclicloess may take a few minutes.
#> Elements of the experimental design in order to check the coding of the data:
#> Tested doses and number of replicates for each dose:
#>
#> 0 0.69 1.223 2.148 3.774 6.631
#> 5 5 5 5 5 5
#> Number of items: 1000
#> Identifiers of the first 20 items:
#> [1] "1" "2" "3" "4" "5.1" "5.2" "6.1" "6.2" "7.1" "7.2"
#> [11] "8.1" "8.2" "9.1" "9.2" "10.1" "10.2" "11.1" "11.2" "12.1" "12.2"
#> Data were normalized between arrays using the following method: cyclicloess
(s_quad <- itemselect(o, select.method = "quadratic", FDR = 0.001))
#> Removing intercept from test coefficients
#> Number of selected items using a quadratic trend test with an FDR of 0.001: 78
#> Identifiers of the first 20 most responsive items:
#> [1] "384.2" "383.1" "383.2" "384.1" "301.1" "363.1" "300.2" "364.2" "364.1"
#> [10] "363.2" "301.2" "300.1" "351.1" "350.2" "239.1" "240.1" "240.2" "370"
#> [19] "15" "350.1"
(f <- drcfit(s_quad, progressbar = TRUE))
#> The fitting may be long if the number of selected items is high.
#>
|
| | 0%
|
|= | 1%
|
|== | 3%
|
|=== | 4%
|
|==== | 5%
|
|==== | 6%
|
|===== | 8%
|
|====== | 9%
|
|======= | 10%
|
|======== | 12%
|
|========= | 13%
|
|========== | 14%
|
|=========== | 15%
|
|============ | 17%
|
|============= | 18%
|
|============= | 19%
|
|============== | 21%
|
|=============== | 22%
|
|================ | 23%
|
|================= | 24%
|
|================== | 26%
|
|=================== | 27%
|
|==================== | 28%
|
|===================== | 29%
|
|====================== | 31%
|
|====================== | 32%
|
|======================= | 33%
|
|======================== | 35%
|
|========================= | 36%
|
|========================== | 37%
|
|=========================== | 38%
|
|============================ | 40%
|
|============================= | 41%
|
|============================== | 42%
|
|=============================== | 44%
|
|=============================== | 45%
|
|================================ | 46%
|
|================================= | 47%
|
|================================== | 49%
|
|=================================== | 50%
|
|==================================== | 51%
|
|===================================== | 53%
|
|====================================== | 54%
|
|======================================= | 55%
|
|======================================= | 56%
|
|======================================== | 58%
|
|========================================= | 59%
|
|========================================== | 60%
|
|=========================================== | 62%
|
|============================================ | 63%
|
|============================================= | 64%
|
|============================================== | 65%
|
|=============================================== | 67%
|
|================================================ | 68%
|
|================================================ | 69%
|
|================================================= | 71%
|
|================================================== | 72%
|
|=================================================== | 73%
|
|==================================================== | 74%
|
|===================================================== | 76%
|
|====================================================== | 77%
|
|======================================================= | 78%
|
|======================================================== | 79%
|
|========================================================= | 81%
|
|========================================================= | 82%
|
|========================================================== | 83%
|
|=========================================================== | 85%
|
|============================================================ | 86%
|
|============================================================= | 87%
|
|============================================================== | 88%
|
|=============================================================== | 90%
|
|================================================================ | 91%
|
|================================================================= | 92%
|
|================================================================== | 94%
|
|================================================================== | 95%
|
|=================================================================== | 96%
|
|==================================================================== | 97%
|
|===================================================================== | 99%
|
|======================================================================| 100%
#> Results of the fitting using the AICc to select the best fit model
#> 11 dose-response curves out of 78 previously selected were removed
#> because no model could be fitted reliably.
#> Distribution of the chosen models among the 67 fitted dose-response curves:
#>
#> Hill linear exponential Gauss-probit
#> 0 11 30 23
#> log-Gauss-probit
#> 3
#> Distribution of the trends (curve shapes) among the 67 fitted dose-response curves:
#>
#> U bell dec inc
#> 6 20 22 19
(r <- bmdcalc(f))
#> 1 BMD-xfold values and 0 BMD-zSD values are not defined (coded NaN as
#> the BMR stands outside the range of response values defined by the model).
#> 28 BMD-xfold values and 0 BMD-zSD values could not be calculated (coded
#> NA as the BMR stands within the range of response values defined by the
#> model but outside the range of tested doses).
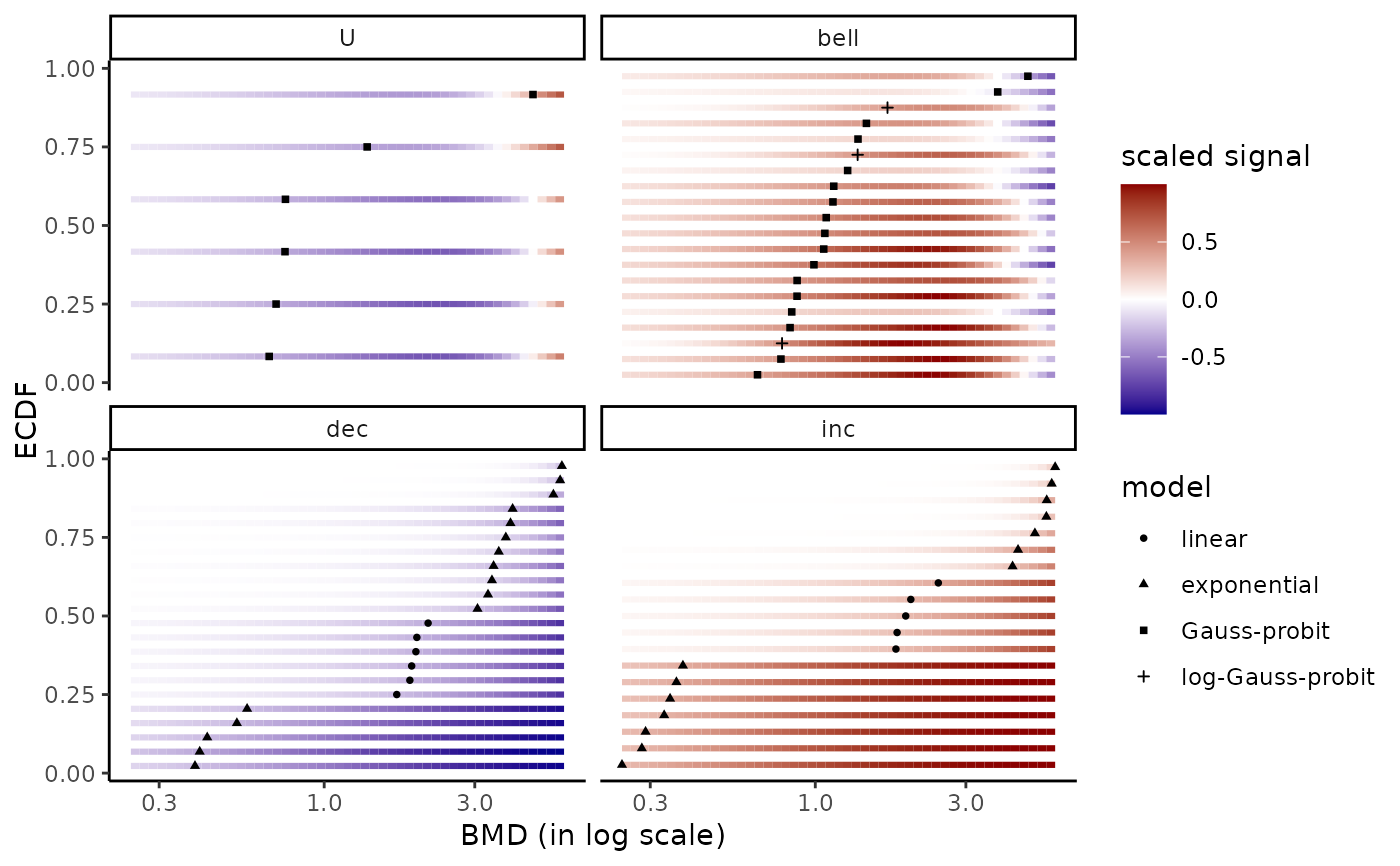
bmdplotwithgradient(r$res, BMDtype = "zSD",
facetby = "trend",
shapeby = "model")
#> Warning:
#> By default xmax was fixed at the maximal BMD value, but you could also
#> fix it at the maximal tested dose.
 # without scaling
bmdplotwithgradient(r$res, BMDtype = "zSD",
scaling = FALSE, facetby = "trend",
shapeby = "model")
#> Warning:
#> By default xmax was fixed at the maximal BMD value, but you could also
#> fix it at the maximal tested dose.
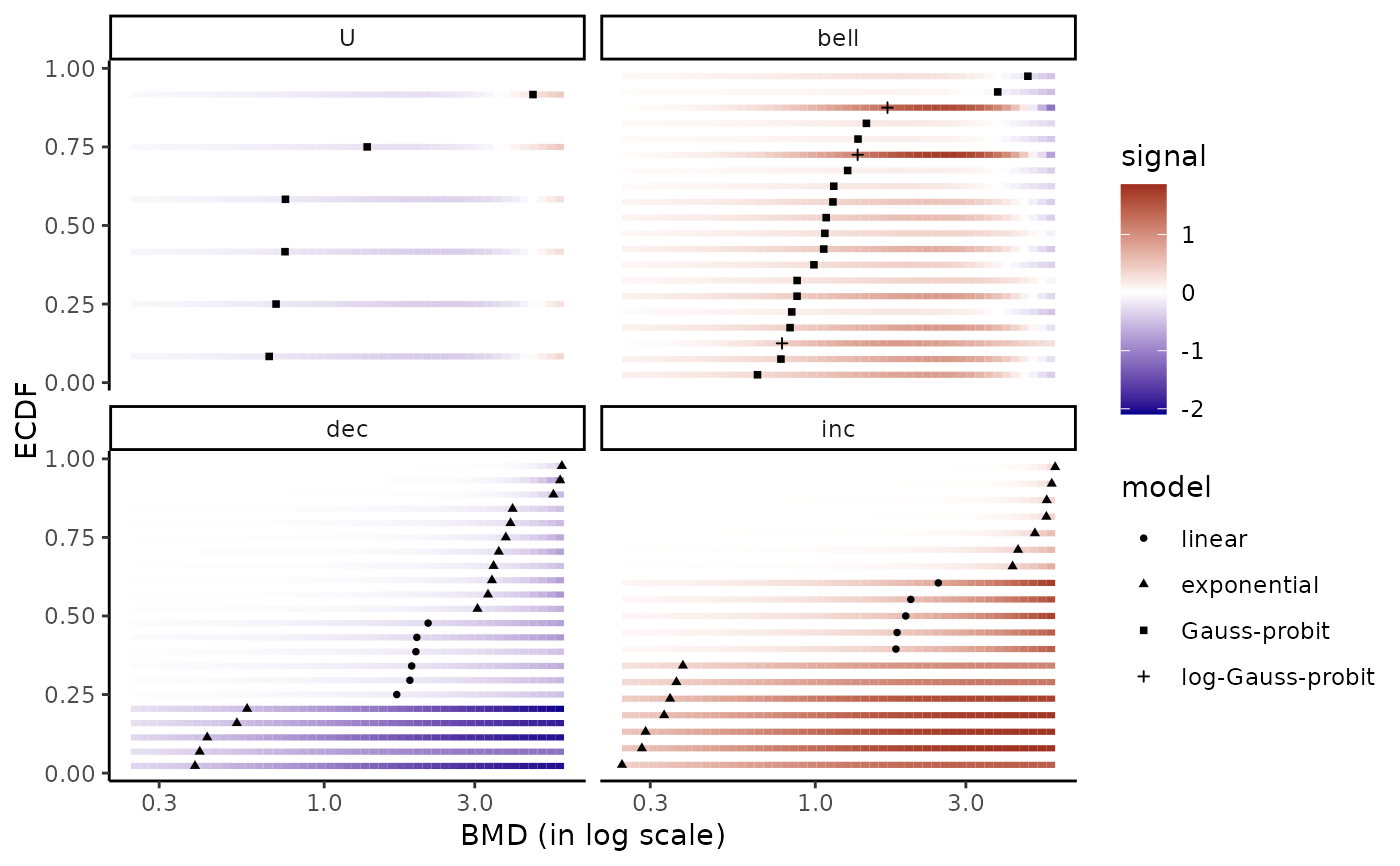
# without scaling
bmdplotwithgradient(r$res, BMDtype = "zSD",
scaling = FALSE, facetby = "trend",
shapeby = "model")
#> Warning:
#> By default xmax was fixed at the maximal BMD value, but you could also
#> fix it at the maximal tested dose.
 # }
# }