Import, check and normalization of single-channel microarray data
microarraydata.RdSingle-channel microarray data in log2 are imported from a .txt file
(internally imported using the function read.table),
checked or from a R object of class data.frame
(see the description
of argument file for the required format
of data)and normalized (between arrays
normalization).
omicdata is a deprecated version of microarraydata.
Usage
microarraydata(file, backgrounddose, check = TRUE,
norm.method = c("cyclicloess", "quantile", "scale", "none"))
omicdata(file, backgrounddose, check = TRUE,
norm.method = c("cyclicloess", "quantile", "scale", "none"))
# S3 method for class 'microarraydata'
print(x, ...)
# S3 method for class 'microarraydata'
plot(x, range4boxplot = 0, ...)Arguments
- file
The name of the .txt file (e.g.
"mydata.txt") containing one row per item, with the first column corresponding to the identifier of each item, and the other columns giving the responses of the item for each replicate at each dose or concentration. In the first line, after a name for the identifier column, we must have the tested doses or concentrations in a numeric format for the corresponding replicate (for example, if there are triplicates for each treatment, the first line could be "item", 0, 0, 0, 0.1, 0.1, 0.1, etc.). This file is imported within the function using the functionread.tablewith its default field separator (sep argument) and its default decimal separator (dec argument at "."). Alternatively an R object of classdata.framecan be directly given in input, corresponding to the output ofread.table(file, header = FALSE)on a file described as above.- backgrounddose
This argument must be used when there is no dose at zero in the data, to prevent the calculation of the BMD by extrapolation. All doses below or equal to the value given in backgrounddose will be fixed at 0, so as to be considered at the background level of exposition.
- check
If TRUE the format of the input file is checked.
- norm.method
If
"none"no normalization is performed, else a normalization is performed using the functionnormalizeBetweenArraysof thelimmapackage using the specified method.- x
An object of class
"microarraydata".- range4boxplot
An argument passed to boxplot(), fixed by default at 0 to prevent the producing of very large plot files due to many outliers. Can be put at 1.5 to obtain more classical boxplots.
- ...
further arguments passed to print or plot functions.
Details
This function imports the data, checks their format (see the description
of argument file for the required format
of data) and gives in the print information
that should help the user to check that the coding of data is correct : the tested doses (or concentrations)
the number of replicates for each dose, the number of items, the identifiers
of the first 20 items. If the argument norm.method is not "none",
data are normalized using the function normalizeBetweenArrays of the
limma package using the specified method : "cyclicloess" (default choice),
"quantile" or "scale".
Value
microarraydata returns an object of class "microarraydata", a list with 9 components:
- data
the numeric matrix of normalized responses of each item in each replicate (one line per item, one column per replicate)
- dose
the numeric vector of the tested doses or concentrations corresponding to each column of data
- item
the character vector of the identifiers of the items, corresponding to each line of data
- design
a table with the experimental design (tested doses and number of replicates for each dose) for control by the user
- data.mean
the numeric matrix of mean responses of each item per dose (mean of the corresponding replicates) (one line per item, one column per unique value of the dose
- data.sd
the numeric matrix of standard deviations of the response of each item per dose (sd of the corresponding replicates, NA if no replicate) (one line per item, one column per unique value of the dose)
- norm.method
The normalization method specified in input
- data.beforenorm
the numeric matrix of responses of each item in each replicate (one line per item, one column per replicate) before normalization
- containsNA
always at FALSE as microarray data are not allowed to contain NA values
The print of a microarraydata object gives the tested doses (or concentrations)
and number of replicates for each dose, the number of items, the identifiers
of the first 20 items (for check of good coding of data) and the normalization method.
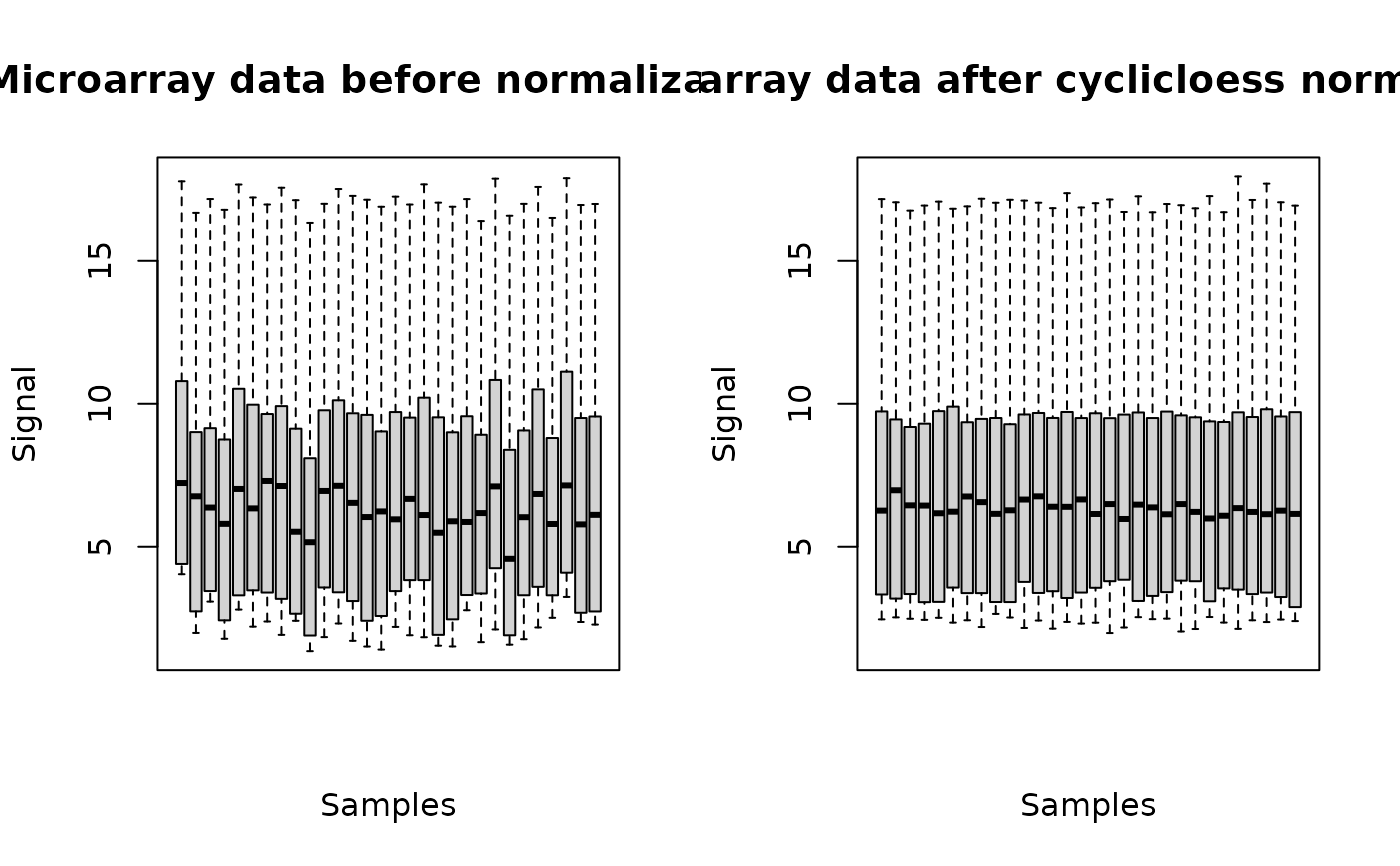
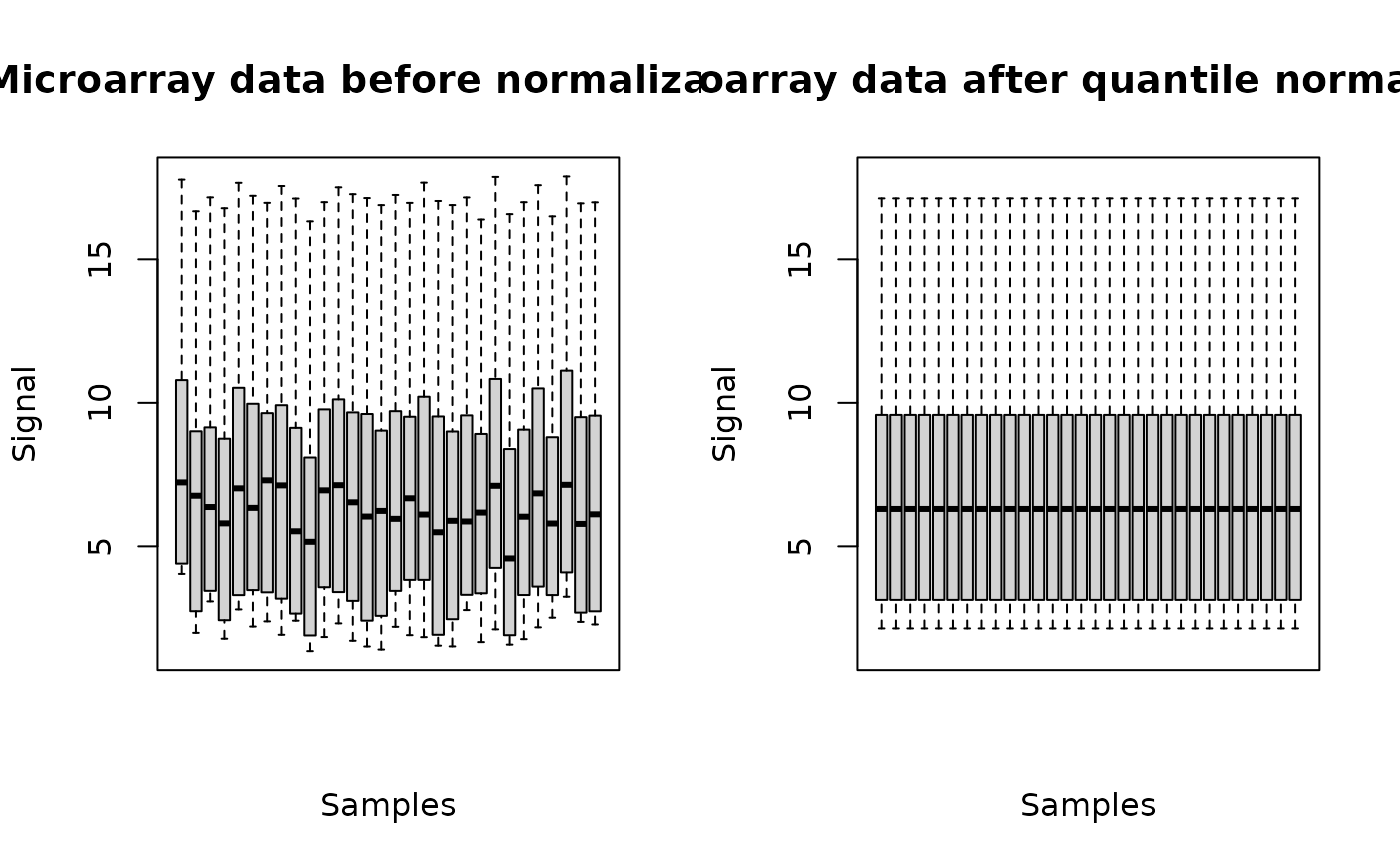
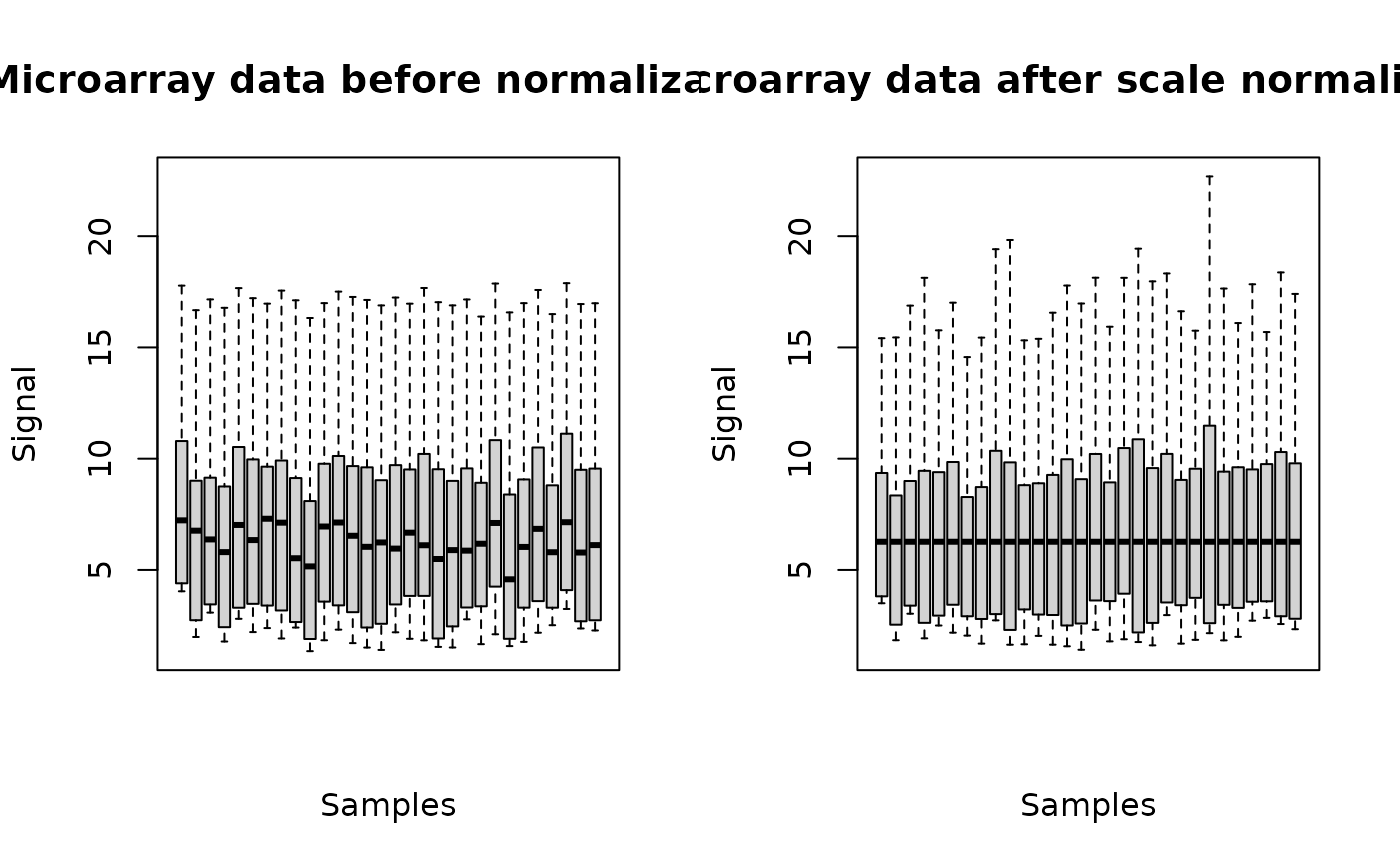
The plot of a microarraydata object shows the data distribution for each dose or concentration and replicate before and after normalization.
See also
See read.table the function used to import data,
normalizeBetweenArrays for details about the normalization and
RNAseqdata, continuousomicdata and
continuousanchoringdata for other types of data.
References
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth, GK (2015), limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Research 43, e47.
Examples
# (1) import, check and normalization of microarray data
# (an example on a subsample of a greater data set published in Larras et al. 2018
# Transcriptomic effect of triclosan in the chlorophyte Scenedesmus vacuolatus)
datafilename <- system.file("extdata", "transcripto_very_small_sample.txt",
package="DRomics")
o <- microarraydata(datafilename, check = TRUE, norm.method = "cyclicloess")
#> Just wait, the normalization using cyclicloess may take a few minutes.
print(o)
#> Elements of the experimental design in order to check the coding of the data:
#> Tested doses and number of replicates for each dose:
#>
#> 0 0.69 1.223 2.148 3.774 6.631
#> 5 5 5 5 5 5
#> Number of items: 100
#> Identifiers of the first 20 items:
#> [1] "1" "2" "3" "4" "5.1" "6.1" "7.1" "8.1" "9.1" "10.1"
#> [11] "11.1" "12.1" "13.1" "14.1" "15" "16.1" "17.1" "18.1" "19.1" "20.1"
#> Data were normalized between arrays using the following method: cyclicloess
plot(o)
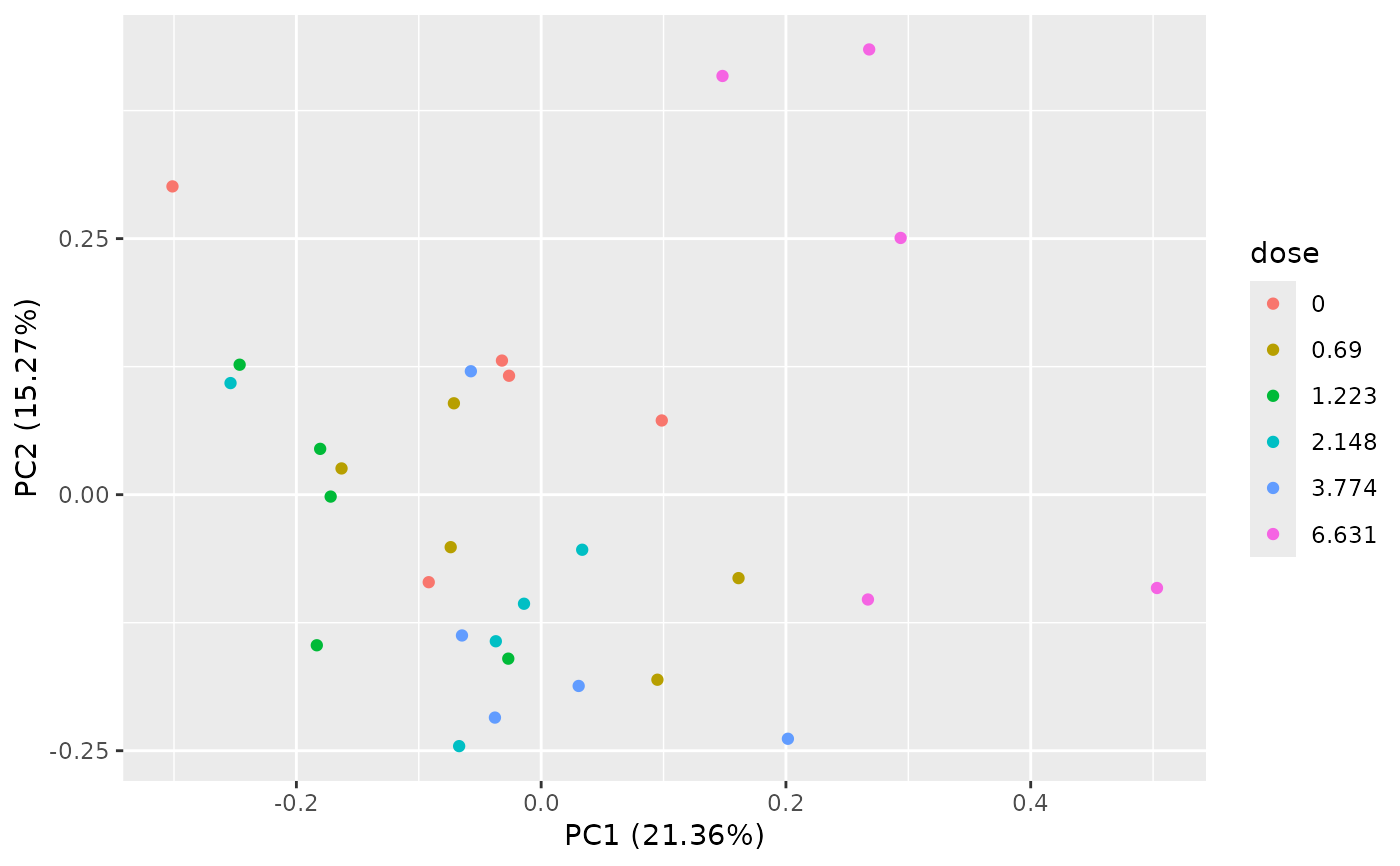
 PCAdataplot(o)
PCAdataplot(o)
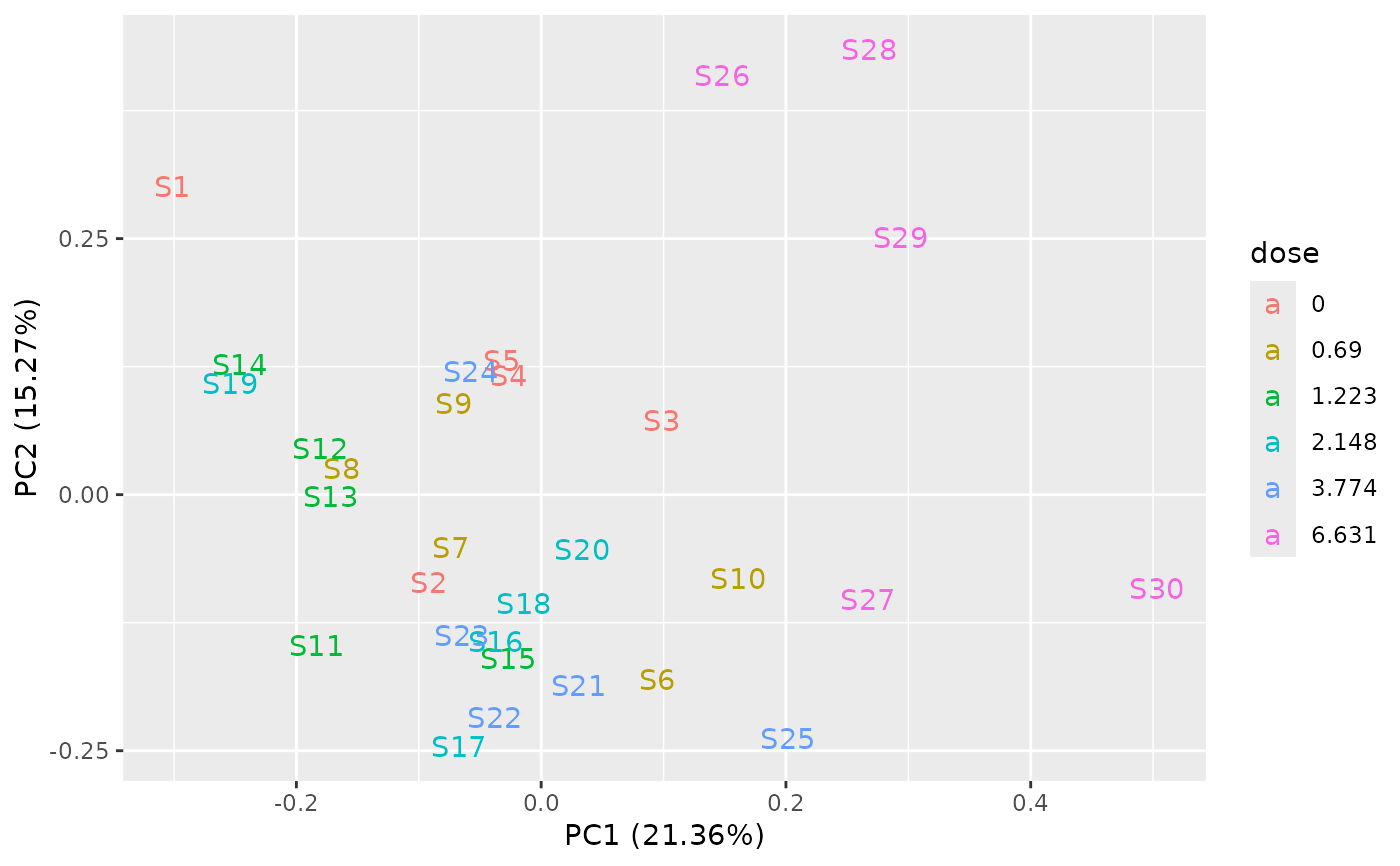
 PCAdataplot(o, label = TRUE)
PCAdataplot(o, label = TRUE)
 # If you want to use your own data set just replace datafilename,
# the first argument of microarraydata(),
# by the name of your data file (e.g. "mydata.txt")
#
# You should take care that the field separator of this data file is one
# of the default field separators recognised by the read.table() function
# when it is used with its default field separator (sep argument)
# Tabs are recommended.
# \donttest{
# (2) normalization with other methods
(o.2 <- microarraydata(datafilename, check = TRUE, norm.method = "quantile"))
#> Elements of the experimental design in order to check the coding of the data:
#> Tested doses and number of replicates for each dose:
#>
#> 0 0.69 1.223 2.148 3.774 6.631
#> 5 5 5 5 5 5
#> Number of items: 100
#> Identifiers of the first 20 items:
#> [1] "1" "2" "3" "4" "5.1" "6.1" "7.1" "8.1" "9.1" "10.1"
#> [11] "11.1" "12.1" "13.1" "14.1" "15" "16.1" "17.1" "18.1" "19.1" "20.1"
#> Data were normalized between arrays using the following method: quantile
plot(o.2)
# If you want to use your own data set just replace datafilename,
# the first argument of microarraydata(),
# by the name of your data file (e.g. "mydata.txt")
#
# You should take care that the field separator of this data file is one
# of the default field separators recognised by the read.table() function
# when it is used with its default field separator (sep argument)
# Tabs are recommended.
# \donttest{
# (2) normalization with other methods
(o.2 <- microarraydata(datafilename, check = TRUE, norm.method = "quantile"))
#> Elements of the experimental design in order to check the coding of the data:
#> Tested doses and number of replicates for each dose:
#>
#> 0 0.69 1.223 2.148 3.774 6.631
#> 5 5 5 5 5 5
#> Number of items: 100
#> Identifiers of the first 20 items:
#> [1] "1" "2" "3" "4" "5.1" "6.1" "7.1" "8.1" "9.1" "10.1"
#> [11] "11.1" "12.1" "13.1" "14.1" "15" "16.1" "17.1" "18.1" "19.1" "20.1"
#> Data were normalized between arrays using the following method: quantile
plot(o.2)
 (o.3 <- microarraydata(datafilename, check = TRUE, norm.method = "scale"))
#> Elements of the experimental design in order to check the coding of the data:
#> Tested doses and number of replicates for each dose:
#>
#> 0 0.69 1.223 2.148 3.774 6.631
#> 5 5 5 5 5 5
#> Number of items: 100
#> Identifiers of the first 20 items:
#> [1] "1" "2" "3" "4" "5.1" "6.1" "7.1" "8.1" "9.1" "10.1"
#> [11] "11.1" "12.1" "13.1" "14.1" "15" "16.1" "17.1" "18.1" "19.1" "20.1"
#> Data were normalized between arrays using the following method: scale
plot(o.3)
(o.3 <- microarraydata(datafilename, check = TRUE, norm.method = "scale"))
#> Elements of the experimental design in order to check the coding of the data:
#> Tested doses and number of replicates for each dose:
#>
#> 0 0.69 1.223 2.148 3.774 6.631
#> 5 5 5 5 5 5
#> Number of items: 100
#> Identifiers of the first 20 items:
#> [1] "1" "2" "3" "4" "5.1" "6.1" "7.1" "8.1" "9.1" "10.1"
#> [11] "11.1" "12.1" "13.1" "14.1" "15" "16.1" "17.1" "18.1" "19.1" "20.1"
#> Data were normalized between arrays using the following method: scale
plot(o.3)
 # }
# }